缺氧诱导因子-1α通过调控蛋白激酶B/核因子-κB信号通路促进肝癌细胞干细胞标志物CD133的表达
2022-05-26赵金金崔非非莫清江林志强张海光焦路阳
赵金金,崔非非,莫清江,汪 磊,林志强,张海光,焦路阳
(1.新乡医学院第一附属医院检验科,河南 卫辉 453100;2.新乡医学院第一附属医院妇产科,河南 卫辉 453100)
肝癌是我国常见的恶性肿瘤[1],作为实体瘤其内部通常存在缺氧,而肿瘤组织缺氧时可表达缺氧诱导因子;缺氧诱导因子-1α(hypoxia inducible factor-1α,HIF-1α)是一种主要的缺氧诱导因子,在肿瘤的发生、发展及治疗中起到关键作用[2-5]。近期有研究报道,HIF-1α在多种肿瘤细胞中诱导干细胞的产生[6-7];本课题组先前研究证实,HIF-1α可促进肝癌细胞干细胞增多,从而使肝癌细胞对化学治疗药物表阿霉素耐药[8]。但是HIF-1α在肝癌细胞中诱导干细胞的产生机制仍不清楚。
蛋白激酶B(protein kinase B,PKB/Akt)/核因子-κB(nuclear factor-κB,NF-κB)信号通路活化在多种肿瘤的发生、发展中发挥重要作用,该信号通路与其他通路相互作用共同参与细胞的增殖、分化和凋亡等生物学过程[9]。NF-κB 家族成员能两两结合成同源性或异源性二聚体,最为常见的是 p50/p65 异源二聚体,其能迅速被多种刺激激活[10]。Akt为NF-κB的上游信号,其活化后诱导NF-κB活化,使p65蛋白磷酸化并进入细胞核,可诱导细胞周期素(cycling D1)、原癌基因c-myc、上皮间质转化(epithelial mesenchymal transformation,EMT)相关蛋白的表达,进而促进细胞的增殖、侵袭[11]。目前,Akt/NF-κB信号通路在肝癌干细胞中是否发挥作用研究报道较少。基于此,本研究通过比较HIF-1α高表达与低表达肝癌细胞内Akt及p65的磷酸化水平,探讨Akt/NF-κB信号通路在HIF-1α诱导肝癌干细胞中发挥的作用,并用Akt和p65抑制剂进行验证,以期为基于HIF-1α的肝癌治疗提供更多的实验依据。
1 材料与方法
1.1 细胞、主要试剂与仪器肝癌细胞HepG2购自中国科学院细胞库,HIF-1α高表达慢病毒pHBLV-HIF1A-3flag-ZsGreen-Puro和对照空载体慢病毒pHBLV-ZsGreen-Puro购自上海汉恒生物科技有限公司,嘌呤霉素、青霉素-链霉素双抗、达尔伯克改良伊格尔培养基(Dulbecco′s modified Eagle′s medium,DMEM)培养液、胎牛血清购自美国Sigma公司,磷酸化蛋白激酶B(phosphorylation protein kinase B,p-Akt)、p-p65抗体购自美国Cell Signaling Technology公司,Akt、p65、甘油醛-3-磷酸脱氢酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)抗体购自武汉三鹰生物技术有限公司。CD133-APC流式抗体购自美国Biolegend生物科技公司,二喹啉甲酸(bicinchoninic acid,BCA)蛋白浓度测定试剂盒购自江苏碧云天生物科技有限公司,四甲基偶氮唑盐(methylthia zolyldiphenyl-tetrazolium bromide,MTT)、Akt激酶抑制剂及NF-κB信号通路抑制剂芒果苷购自美国MCE公司,其他所用试剂均为国产分析纯。CO2恒温培养箱及全波长扫描酶标仪购自美国赛默飞世尔科技有限公司,BIO-RAD Mini Trans-Blot垂直电泳系统购自美国伯乐公司,Axio Observer A1倒置荧光显微镜购自德国卡尔蔡司集团,BD Calibur流式细胞仪购自美国碧迪公司,Amersham Imager 6000化学发光荧光成像系统购自美国General Electric Company公司,Countstar智能细胞分析仪购自上海睿钰生物科技有限公司。
1.2 实验方法
1.2.1 细胞培养及慢病毒转染肝癌细胞HepG2用含质量分数1%青霉素-链霉素双抗、体积分数10%胎牛血清的DMEM培养液于含体积分数5% CO2、37 ℃恒温培养箱中培养,当细胞达到90%融合度时用质量分数0.25%胰酶消化并计数,按每孔1×106个细胞铺于6孔板,将细胞随机分为空白对照组、对照组和实验组。按照汉恒生物慢病毒说明书进行转染,首先从冰箱中取出慢病毒融化,吸去细胞原有培养基,加入1 mL含4 mg·L-1聚凝胺的新鲜培养基;空白对照组不加病毒原液,对照组细胞中加入20 μL病毒滴度为2×1011TU·L-1的pHBLV-ZsGreen-Puro病毒原液,实验组细胞中加入20 μL病毒滴度为2×1011TU·L-1的pHBLV-HIF-1α-3flag-ZsGreen-Puro病毒原液;轻轻混匀,第2天吸去含有病毒的培养液,换新鲜培养液继续培养。为得到稳定表达HIF-1α的细胞株HepG2-HIF-1α,在感染72 h后将细胞消化,制成单细胞悬液,以每孔1个细胞的量接种于96孔板,加入嘌呤霉素,每3 d换液1次,3周后消化细胞并扩大培养。
1.2.2 荧光显微镜下观察细胞绿色荧光蛋白(green fluorescent protein,GFP)表达取空白对照组、对照组和实验组生长至60%~80%融合度的细胞,于荧光显微镜下观察细胞,并使用化学发光荧光成像系统采集照片;计数绿色荧光下细胞数量和同一视野白光下细胞的数量,计算GFP阳性细胞比例,GFP阳性细胞比例=绿色荧光下细胞的数量/白光下细胞数量×100%。
1.2.3 Western blot检测HIF-1α蛋白相对表达量及Akt/NF-κB信号通路蛋白p-Akt/Akt、p-p65/p65蛋白表达取空白对照组、对照组和实验组细胞贴壁生长过夜,第2天用冷的PBS洗3次,加入细胞裂解液冰上孵育20 min,12 000×g离心15 min,收集上清得到细胞裂解液。BCA试剂盒测定细胞裂解液中蛋白浓度,用细胞裂解液将3组细胞调至相同浓度;加入上样缓冲液。每个样品上样量为30 μg,采用变性十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodiumdodecylsulfate-polyacrylamide gel electrophoresis,SDS-PAGE)进行电泳,将蛋白转印至聚偏二氟乙烯膜,质量分数5%脱脂奶粉封闭2 h,加入HIF-1α、p-Akt、p-p65、Akt、p65一抗(滴度均为11 000)和GAPDH一抗(滴度为110 000),4 ℃孵育过夜,Tris 缓冲生理盐水( Tris-buffered saline and Tween-20,TBST)洗涤5次,每次5 min;加入辣根过氧化物酶标记的抗鼠或抗兔二抗(滴度为110 000),室温孵育1 h,TBST洗涤5次,每次5 min;用增强化学发光试剂显色,应用Amersham Imager 600凝胶成像系统拍照,Image J软件分析蛋白表达量,HIF-1α、p-Akt、Akt、p-p65、p65蛋白相对表达量以HIF-1α、p-Akt、Akt、p-p65、p65蛋白灰度值与GAPDH灰度值比值表示,计算p-Akt/Akt、p-p65/p65的比值。实验重复3次,取均值。
1.2.4 流式细胞术检测细胞CD133的表达取对照组和实验组生长至90%融合度的细胞,PBS冲洗后,加入质量分数0.25%的胰酶溶液消化细胞,使用Countstar智能细胞分析仪进行细胞计数,取1×106个细胞PBS洗涤后加入100 μL APC标记的CD133抗体(滴度为1100),避光染色15 min,PBS洗涤,然后加入200 μL PBS混匀,应用流式细胞仪检测CD133表达,应用Flowjo软件进行分析并计算CD133阳性细胞率。
1.2.5 Akt和p65抑制剂干预细胞的细胞活力和CD133表达的检测取实验组生长至90%融合度的HepG2-HIF-1α细胞,按照“1.2.4”项方法消化细胞并计数,按每孔1×104个细胞接种于96孔板,随机分为HepG2-HIF-1α组、Akt激酶抑制剂组和NF-κB信号通路抑制剂组。第2天,Akt激酶抑制剂组加入终浓度为0.4、0.8、1.6、3.2、6.3、12.5、25.0 μmol·L-1的Akt激酶抑制剂,NF-κB信号通路抑制剂组分别加入终浓度为3.0、6.0、12.5、25.0、50.0、100.0、200.0、400.0 μmol·L-1NF-κB信号通路抑制剂,设置空白对照组,继续培养24 h;每孔加入20 μL质量浓度为5 g·L-1的MTT溶液,于培养箱中孵育2 h,吸弃培养液,加入200 μL 二甲基亚砜溶解10 min,于570 nm波长处测定吸光度值,计算细胞活力,细胞活力=不同浓度处理组细胞吸光值/空白对照组细胞吸光值。实验重复3次,取均值。另取HepG2-HIF-1α组、AKT激酶抑制剂组和NF-κB信号通路抑制剂组细胞,按“1.2.4”项方法检测细胞中CD133的表达。

2 结果
2.1 空白对照组、对照组和实验组GFP阳性细胞率及HIF-1α蛋白相对表达量比较结果见表1和图1、图2。对照组和实验组GFP阳性细胞率显著高于空白对照组,差异有统计学意义(P<0.05);对照组与实验组GFP阳性细胞率比较差异无统计学意义(P>0.05)。实验组细胞中HIF-1α蛋白相对表达量显著高于空白对照组及对照组,差异有统计学意义(P<0.05);空白对照组与对照组细胞中HIF-1α蛋白相对表达量比较差异无统计学意义(P>0.05)。
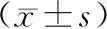
表1 空白对照组、对照组和实验组GFP阳性细胞率及细胞中HIF-1α蛋白相对表达量比较
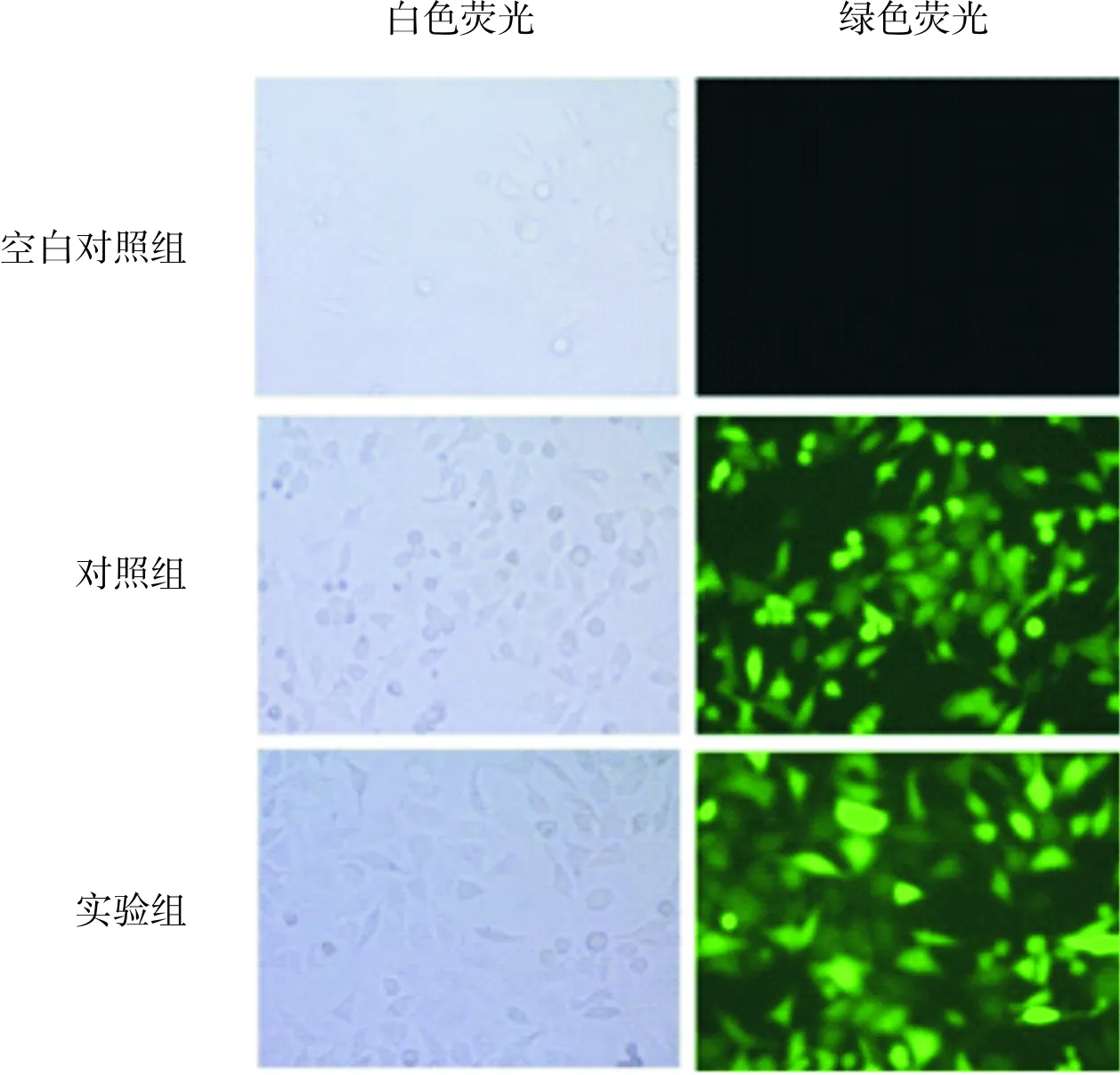
图1 空白对照组、对照组和实验组细胞蛋白GFP蛋白表达(×200)

1:空白对照组;2:对照组;3:实验组。
2.2 对照组和实验组细胞中p-Akt/Akt、p-p65/p65比较结果见表2和图3。实验组细胞中p-Akt/Akt、p-p65/p65蛋白表达水平显著高于对照组,差异有统计学意义(P<0.05)。
表2 对照组和实验组细胞中p-Akt/Akt、p-p65/p65比较
1:对照组;2:实验组。
2.3 对照组和实验组CD133阳性细胞率比较结果见图4。对照组、实验组CD133阳性细胞率分别为(4.42±0.29)%、(15.43±0.41)%,实验组CD133阳性细胞率显著高于对照组,差异有统计学意义(t=22.160,P<0.05)。

图4 流式细胞仪检测对照组和实验组细胞中CD133的表达
2.4 Akt激酶抑制剂和NF-κB信号通路抑制剂干预细胞的细胞活力和CD133表达的比较0.4、0.8、1.6、3.2、6.3、12.5、25.0 μmol·L-1Akt激酶抑制剂组细胞活力分别为0.920±0.076、0.833±0.066、0.854±0.082、0.915±0.153、0.913±0.110、0.843±0.039、0.927±0.049,各浓度组间细胞活力比较差异无统计学意义(F=1.301,P>0.05);3.0、6.0、12.5、25.0、50.0、100.0、200.0、400.0 μmol·L-1NF-κB信号通路抑制剂组细胞活力分别为0.978±0.065、0.894±0.050、0.995±0.028、0.987±0.043、0.982±0.067、1.055±0.112、0.901±0.030、0.937±0.056,各浓度组间细胞活力比较差异无统计学意义(F=2.300,P>0.05)。HepG2-HIF-1α组、Akt激酶抑制剂组和NF-κB信号通路抑制剂组CD133阳性细胞率分别为(15.43±0.41)%、(8.61±0.53)%、(9.86±0.47)%,3组CD133阳性细胞率比较差异有统计学意义(F=171.9,P<0.05);Akt激酶抑制剂组和NF-κB信号通路抑制剂组CD133阳性细胞率显著低于HepG2-HIF-1α组,差异有统计学意义(t=10.170、8.932,P<0.05);Akt激酶抑制剂组与NF-κB信号通路抑制剂组CD133阳性细胞率比较差异无统计学意义(t=1.745,P>0.05)。
3 讨论
肝癌作为一种实体肿瘤,肿瘤内通常存在缺氧,此时HIF-1α的降解过程受到抑制,细胞中大量表达HIF-1α[11-12]。HIF-1α作为一种转录因子调控细胞内胰岛素、胰岛素样生长因子、表皮生长因子等多种蛋白的表达,对细胞的增殖、迁移、凋亡及干细胞特性等发挥重要调控作用[13]。研究结果显示,在乳腺癌中缺氧诱导的HIF-1α可通过A2BR/PKCδ通路诱导乳腺癌干细胞[14]。目前,关于HIF-α对肝癌细胞的干细胞特性影响的相关研究报道较少。因此,本研究通过构建HIF-1α过表达细胞HepG2-HIF-1α,观察HepG2-HIF-1α与HepG2细胞中CD133的表达差异,以及细胞中p-Akt/Akt、p-p65/p65的表达变化,分析HIF-α对肝癌细胞的干细胞特性的影响及机制。
慢病毒过表达载体是生命科学研究中常用的实验方法[15],通常用于细胞或动物内基因的表达与沉默。本研究中将含有GFP标签及嘌呤霉素抗性基因的慢病毒载体经HIF-1A基因导入至肝癌细胞HepG2中,结果显示,对照组和实验组细胞中GFP相对表达量显著高于空白对照组;对照组与实验组细胞中GFP相对表达量比较差异无统计学意义。实验组细胞中HIF-1α蛋白相对表达量显著高于空白对照组及对照组;空白对照组与对照组细胞中HIF-1α蛋白相对表达量比较差异无统计学意义。该结果证明,含有HIF-1α基因的慢病毒成功转染至HepG2细胞中,并能够正常表达,HepG2-HIF-1α细胞构建成功。
肿瘤干细胞是指在肿瘤组织内具有自我更新和分化增殖潜能的细胞[16],在肿瘤的发生、发展及治疗过程中发挥重要作用[17]。国内外研究已证实,包括肝癌在内的多种实体肿瘤中存在肿瘤干细胞,干细胞的存在可能是肿瘤治疗失败的一个主要原因。通常用作肝癌干细胞的标志物有CD13、CD24、CD44、CD90、CD133、OCT4、EpCAM等[18]。CD133是一种独特的干细胞标志物[19-22],相对分子质量为120 000,是一个由大约850个氨基酸组成的多肽链,具有5个跨膜结构域和2个巨大的胞外环,胞外环上具有4个潜在的糖基化位点,这些位点可与靶分子的结合位点结合而发挥调控作用。CD133在肿瘤细胞干性及肿瘤耐药方面发挥重要作用,日本学者WAKIZAKA等[18]研究报道,干细胞标志物CD133或EpCAM的高表达与肝癌患者的生存期缩短相关。LIU等[21]报道,CD133与肝癌细胞的化学治疗的抵抗性、放射治疗的抵抗性及代谢重编程相关。本研究结果显示,实验组CD133阳性细胞率显著高于对照组,证实HIF-α蛋白过表达可增加肝癌细胞系HepG2的干细胞特性。
CD133调控机制已成为近年来的研究热点,研究发现,Wnt/β-catenin、Hedgehog及Stat3信号通路均可促进CD133表达,进而导致肝癌细胞持续自我更新[23-24]。Akt在细胞存活和凋亡中起重要作用,近年来研究发现,Akt信号通路与肝癌细胞的自我更新及干细胞特性相关;CDCA8可通过抑制Akt/β-actenin 信号通路抑制肝癌细胞干细胞特性[25];HYD-PEP06可通过抑制Akt 信号通路抑制肝癌干细胞特性[26];KAHRAMAN等[27]研究显示,IL-8可通过PI3K/Akt/mTOR信号通路诱导肝癌干细胞特性。细胞内多种蛋白及miRNA的异常表达可通过活化Akt信号通路使肝癌细胞具有干细胞特性[28-29]。HE等[30]研究发现,在肿瘤细胞中NF-κB信号通路通常异常活化。HU等[31]研究证实,CD13可通过NF-κB信号通路使肝癌细胞具有干细胞的特性。本研究结果显示,实验组细胞中p-Akt/Akt、p-p65/p65显著高于对照组,说明肝癌细胞高表达HIF-1α后Akt/NF-κB信号通路被激活。此外,本研究结果显示,不同浓度的AKT激酶抑制剂组和NF-κB 信号通路抑制剂组HepG2-HIF-1α细胞活力差异均无统计学意义,Akt激酶抑制剂组和NF-κB信号通路抑制剂组CD133阳性细胞率均显著低于HepG2-HIF-1α组;该结果提示,Akt/NF-κB信号通路异常活化对肝癌细胞活力无显著影响,而对肝癌细胞的干细胞特性发挥重要的调控作用,HIF-1α 通过促进Akt及NF-κB的磷酸化激活Akt/NF-κB 信号通路来增加肝癌细胞的干细胞特性。
综上所述,HIF-1α可通过促进Akt及NF-κB p65的磷酸化诱导肝癌细胞表面CD133的表达,增加肝癌细胞的干细胞特性。但是,本研究未检测AKT/NF-κB信号通路中的其他信号蛋白,而检测其他信号蛋白将更有利于阐明HIF-1α诱导肝癌干细胞的特性的机制,后期需进一步研究,以其为以HIF-1α为靶点治疗肝癌提供更多理论证据。
