基于1,3-环己二酮砌块的吲哚类似物合成研究
2022-05-20席爽,王锐
席 爽,王 锐
(辽宁石油化工大学 石油化工学院,辽宁 抚顺 113001)
Japp-Klingemann费舍尔吲哚合成法是Japp-Klingemann反应和费舍尔吲哚环化反应的结合体[1],即通过苯基重氮盐和活泼的亚甲基化合物(β⁃酮酸、β⁃二酮、β⁃酮酸酯等)的烯醇式异构体,在酸性或碱性介质中进行偶联生成芳腙类化合物,随后发生费舍尔吲哚环化反应生成多取代吲哚结构。该反应涉及到芳基重氮阳离子对活泼的亚甲基化合物烯醇式碳原子的亲电攻击,给出一个偶氮中间体;随后,水解生成芳基腙,芳基腙互变异构生成乙烯基肼,然后,经历[3,3]-σ重排、环化生成吲哚类似物;紧接着消除一分子的铵根离子得到吲哚衍生物。1948年,化学家S.P.Findlay等[2]对Japp-Klingemann费舍尔吲哚合成反应进行了报道,该反应可以一锅法实现,也可分步完成。
近年来,1,3-环己二酮及其衍生物作为高效合成砌块在有机合成化学中得到快速发展及广泛应用[3-5]。较为经典的应用是将其整体作为合成砌块应用到化学合成中,从而引入环状结构[6]。此外,利用1,3-环己二酮及其衍生物的碎裂化开环反应得到更有价值的结构单元也受到化学家们的青睐。2016年,Y.F.Tang课题组完成了三环细胞松弛素periconiasins A⁃C的全合成工作[7]。从2-甲基-2-((2E,4E)-4-甲基环己基-2,4-二烯-1-甲基)环己烷-1,3-二酮出发,通过对其酮羰基进行还原去对称和羟基官能团保护,随后羟醛缩合/Grob碎裂化串联反应构建链状聚酮-氨基酸前体,最终实现了三环细胞松弛素periconiasins A⁃C的全合成。合成该类多烯链状产物的传统方法步骤都相对冗长,Y.F.Tang课题组的研究很好地体现了1,3-环己二酮碎裂化开环战略的优势。2015年,J.Rodriguez课题组开发了一种快速合成富对映选择性中环内酯(8~11元环)的新型合成方法[8]。
本文的设计理念是将2-位单侧链取代的1,3-环己二酮作为合成砌块,采用经典的Japp-Klingemann费舍尔吲哚合成法,高效构建2,3-位多取代的吲哚结构单元,这一设计理念未有过相关报道。在传统的Japp-Klingemann费舍尔吲哚合成中,链状β⁃酮酯类化合物在反应中会脱除一分子的羧酸,最终得到吲哚2-位上单羧酸酯取代的吲哚衍生物,而1,3-环己二酮是一个环状的亚甲基化合物,其在反应中接受芳基重氮阳离子的亲电攻击后发生开环反应,在使用不同溶剂的条件下生成的羧酸或羧酸酯官能团仍然会保留在所生成的吲哚衍生物中,官能团的保留可为下游反应创造有力的条件。对该反应进行方法学的条件优化及筛选,通过1,3-环己二酮的碎裂化开环和得到的苯腙结构的重新环合,可在吲哚2-位侧链上引入两个羰基结构,并将其应用于后续吲哚生物碱分子的合成中[9-15]。
1 实验部分
1.1 试剂与仪器
试剂:苯胺(体积分数为99.5%),萨恩化学技术(上海)有限公司;四氟硼酸(质量分数为48.0%)、亚硝基叔丁酯(体积分数为90.0%)、2-甲基-1,3-环己二酮(质量分数为98.0%)、1,3-环己二酮(质量分数为97.0%)、2,6-二甲基-1,4-二氢吡啶-3,5-二甲酸二叔丁酯(质量分数为97.0%),氘代氯仿(氘代氯仿的质量分数为99.8%,三甲基硅烷(TMS)的质量分数为0.03%)、氘代二甲基亚砜(氘代二甲基亚砜的质量分数为99.8%,三甲基硅烷(TMS)的质量分数为0.03%),北京百灵威科技有限公司;苯甲醛(体积分数为99.0%),上海阿拉丁生化科技股份有限公司;L-脯氨酸(质量分数为99.0%),上海皓鸿生物医药科技有限公司;二氯甲烷(分析纯,体积分数为99.5%,含50~150μg/g的异戊烯稳定剂)、乙酸乙酯(体积分数为99.9%),北京伊诺凯科技有限公司。
除非特殊说明,所有反应均在氮气保护且无水条件下进行,一般反应试剂购买后无需任何处理直接使用,所有化学试剂均为商业购得。
仪器:Bruker AV 400型核磁共振仪,瑞士Bruker公司;薄层色谱硅胶板(GF254),青岛海洋化工有限公司;RE-5002旋转蒸发仪,瑞德仪器设备有限公司;层析柱,重庆欣维尔玻璃有限公司。
显色手段包括紫外光照射(紫外波长254 nm及365 nm)、高锰酸钾(加热)、磷钼酸(加热)和碘熏。核磁谱图1H-NMR和13C-NMR采用Bruker AV 400型核磁共振仪测定。所使用的氘代溶剂包括氘代氯仿和氘代二甲基亚砜。氘代氯仿内残留的氯仿作为内标使用(核磁氢谱的化学位移为7.26,核磁碳谱的化学位移为77.16);氘代二甲基亚砜内残留的二甲基亚砜作为内标使用(核磁氢谱的化学位移为2.50,核磁碳谱的化学位移为39.52)。单峰及多重峰的表示方法:单峰=s,双重峰=d,三重峰=t,四重峰=q,宽峰=b,五重峰及以上峰=m。
1.2 实验方法
化合物1的合成:量取苯胺(0.18 mL)于10 mL圆底烧瓶中,加入3.00 mL无水乙醇溶液,并加入大小适中的磁力搅拌子,将圆底烧瓶固定在磁力搅拌器上,重复3次抽换气步骤进行氮气保护,保持瓶中无水无氧状态。将上述溶液冷却至0℃,向该体系中缓慢滴加四氟硼酸(0.34 mL),滴加完成后在0℃下继续搅拌10.0 min。随后缓慢向该体系中缓慢滴加亚硝基叔丁酯(0.31 mL),继续在0℃下搅拌10.0 min,然后移至室温下搅拌1.0 h。用TLC监测反应完全后,向反应体系中缓慢加入乙醚溶液,析出白色盐状沉淀物,将乙醚溶液滤除,随后再用10.00 mL的乙醚溶液洗涤沉淀2次,在减压条件下干燥,得到0.73 g苯基四氟硼酸重氮盐化合物1,产率为95%。化合物1置于-20℃的冰箱中暂存。化合物1的合成路线如图1所示。

图1 化合物1的合成路线
化合物2的合成:称取所制备的化合物1(0.20 g)和2-甲基-1,3-环己二酮化合物(0.40g)于10 mL圆底烧瓶中,再加入5.00 mL无水甲醇溶液,并加入大小适中的干燥的磁力搅拌子,将圆底烧瓶固定在磁力搅拌器上,重复3次抽换气步骤,保持瓶中无水无氧状态。将上述反应液置于室温条件下搅拌3.0 h。将溶液减压浓缩干燥除去甲醇溶剂,加入水10.00 mL,再用10.00 mL的乙酸乙酯洗涤水相3次,合并有机相,无水硫酸钠干燥,抽滤,减压浓缩有机相,经柱层析分离纯化得到0.23 g化合物2,产率为83%。化合物2的合成路线如图2所示。

图2 化合物2的合成路线
化合物3的合成:以化合物2作为模型底物,针对该类底物的费舍尔吲哚关环条件进行了优化筛选。化合物3的合成路线如图3所示,化合物3的关环反应条件筛选见表1。表中,1.5%盐酸、10.0%盐酸表示盐酸的体积分数分别为1.5%、10.0%,下同。

图3 化合物3的合成路线

表1 化合物3的关环反应条件筛选
化合物4的合成:称取1,3-环己二酮(112.00 mg)、2,6-二甲基-1,4-二氢吡啶-3,5-二甲酸二叔丁酯(253.00 mg)、苯甲醛(371.00 mg)和L-脯氨酸(23.00 mg)于10 mL圆底烧瓶中,再加入3.00 mL二氯甲烷溶液,并加入干燥、大小适中的磁力搅拌子,将圆底烧瓶固定在磁力搅拌器上,室温条件下搅拌24.0 h,用TLC监测反应完全后,将溶液减压浓缩干燥除去二氯甲烷溶剂,经柱层析分离纯化得到化合物4。化合物4的合成路线如图4所示。

图4 化合物4的合成路线
化合物5及6的合成:称取2-苯基环己二酮(0.56 g)、苯基四氟硼酸重氮盐1(0.54 g)于50 mL圆底烧瓶中,加入25.00 mL无水乙醇溶液,并加入大小适中的磁力搅拌子,将圆底烧瓶固定在磁力搅拌器上,重复3次抽换气步骤,保持瓶中无水无氧状态。室温条件下搅拌3.0 h,用TLC监测反应完全后,将溶液减压浓缩干燥除去乙醇溶剂,加入水10.00 mL、再用10.00 mL的乙酸乙酯洗涤水相3次,合并有机相,无水硫酸钠干燥,抽滤,减压浓缩有机相,经柱层析分离纯化得到化合物5(0.28 g),产率为81%。化合物5的合成路线如图5所示。

图5 化合物5的合成路线
化合物6的合成路线如图6所示。

图6 化合物6的合成路线
化合物5及6的关环反应条件见表2。

表2 化合物5及6的关环反应条件
条件1的操作步骤:制备体积分数为10.0%的盐酸乙醇溶液。量取10.00 mL无水乙醇于干燥的50 mL圆底烧瓶中,加入干燥、大小适中的磁力搅拌子,置于冰浴条件下搅拌10.0 min,量取1.57 mL乙酰氯并将其缓慢滴加到冰乙醇中,继续搅拌5.0 min,备用。
称取苯腙乙酯化合物5(61.00 mg),置于干燥的10 mL封管中,加入干燥、大小适中的磁力搅拌子,将制备的体积分数为10.0%盐酸乙醇溶液用塑料滴管转移至封管中,用封口塞密封。在110℃油浴搅拌条件下加热回流40.0 min,反应液由红棕色渐变为淡黄色,用TLC监测反应完全后,将反应液转移至25 mL尖底烧瓶中,减压旋干,加入1.00 mL二氯甲烷2次直至粗品全部蒸干,加入二氯甲烷5.00 mL,用饱和碳酸氢钠水溶液萃取,再用10.00 mL的二氯甲烷洗涤水相3次,合并有机相,无水硫酸钠干燥,抽滤,减压浓缩有机相,经柱层析分离纯化得到化合物6(53.00 mg),产率为91%。
条件2的操作步骤:称取苯腙乙酯化合物5(45.00 mg),置于干燥的10 mL封管中,加入干燥、大小适中的磁力搅拌子,将体积分数30.0%盐酸乙醇溶液用塑料滴管转移至封管中,用封口塞密封。在110℃油浴搅拌条件下加热回流20.0 min,反应液由红棕色渐变为淡黄色,用TLC监测反应完全后,将反应液转移至25 mL尖底烧瓶中,减压旋干,加入2次1.00 mL二氯甲烷直至粗品全部蒸干,加入二氯甲烷5.00 mL,用饱和碳酸氢钠水溶液萃取,再用10.00 mL的二氯甲烷洗涤水相3次,合并有机相,无水硫酸钠干燥,抽滤,减压浓缩有机相,经柱层析分离纯化得到化合物6(42.00 mg),产率为98%。
化合物7的合成:化合物7的合成路线如图7所示,操作步骤同条件2。

图7 化合物7的合成路线
2 结果与讨论
2.1 实验数据分析
以2-甲基-1,3-环己二酮为原料制备苯腙甲酯化合物3,并以化合物3作为原料对费舍尔吲哚关环所需条件(酸、溶剂、温度、时间等)进行了筛选(筛选条件见表1)。结果发现,在回流温度为110℃的条件下,以6当量的对甲苯磺酸作为反应所需的酸试剂,甲苯作为溶剂,在封管加热条件下能以49%的产率得到关环产物3。降低反应温度到80℃时,产率下降到27%,采用多聚磷酸和三氟乙酸等其他有机酸时结果未能得到改善(序号3和序号4),在体积分数1.5%盐酸/醋酸条件下也未能得到所需产物。在以三氟甲磺酸为反应用酸、甲苯为溶剂、温度为100℃的条件下,仅用0.5 h,便顺利地得到了产物,其产率为48%,进一步调整溶剂和温度均未提高产率(表1中序号7和序号8)。值得注意的是,在封管加热条件下,采用体积分数为10.0%的盐酸乙醇溶液回流1.0 h,未反应,原料回收处理。结合费舍尔吲哚关环的机理,对反应过程进行分析。化合物3在酸性条件下底物中的亚胺结构首先异构化为烯胺,其后在加热条件下发生[3,3]-σ重排,进而芳构化生成吲哚,可以认为反应初始阶段化合物3的苯腙结构是相对稳定的,通过酸催化使底物3发生异构化生成乙烯基肼不是一个热力学稳定的过程,若在化合物3的甲基碳上引入一个苯基,使其在酸催化发生异构时能形成稳定的共轭结构,或许反应能够顺利发生。结合可能的机理分析,以2-苄基-1,3-环己二酮为原料制备了化合物5,在体积分数为10.0%的盐酸乙醇溶液、封管加热加压条件下,反应只需40.0 min,便以91%的收率得到了吲哚产物6,提高盐酸乙醇混合液的体积分数到30.0%,产率提高到98%。这一结果验证了可能的机理分析。
2.2 Japp-Klingemann费舍尔吲哚合成机理
Japp-Klingemann费舍尔吲哚合成机理如图8所示。

图8 Japp-Klingemann费舍尔吲哚合成机理
2.3 化合物结构表征
化合物4的核磁共振氢谱及核磁共振碳谱如图9所示。由图9(a)可知,化合物4的核磁共振氢谱数据为:1H-NMR(400 MHz,DMSO):δ7.70(d,J=16.0 Hz,1H),8.31(d,J=3.1 Hz,1H),8.24~8.19(m,1H),7.52~7.48(m,1H),7.26~7.17(m,2H),3.62(s,3H),2.91(t,J=7.3 Hz,2H),2.42(t,J=7.4 Hz,2H),1.97~1.88(m,2H)。由图9(b)可知,化合物4的碳原子化学位移数据为:13C-NMR(100 MHz,DMSO-d)δ194.90,173.37,136.72,133.87,125.44,122.86,121.80,121.44,116.35,112.20,51.35,37.86,32.90,20.27。

图9 化合物4的1H-NMR(DMSO,400 MHz)及13C-NMR(DMSO,100 MHz)
化合物6的核磁共振氢谱及核磁共振碳谱如图10所示。由图10(a)可以看出,化合物6的核磁共振氢谱数据为:1H-NMR(400 MHz,CDCl3):δ9.51(s,1H),7.46(s,7H),7.40~7.33(m,1H),4.06(q,J=7.1 Hz,2H),2.54(t,J=7.2 Hz,2H),2.17(t,J=7.5 Hz,2H),1.92(dd,J=14.7,7.4 Hz,2H),1.92(dd,J=14.7,7.4 Hz,2H)。由图10(b)可以看出,化合物6碳原子的化学位移数据为:13C-NMR(100 MHz,CDCl3)δ193.47,173.13,135.85,134.39,131.71,130.63,128.97,128.66,128.09,126.71,124.41,122.24,120.96,112.02,60.39,39.29,33.59,19.86,14.33。
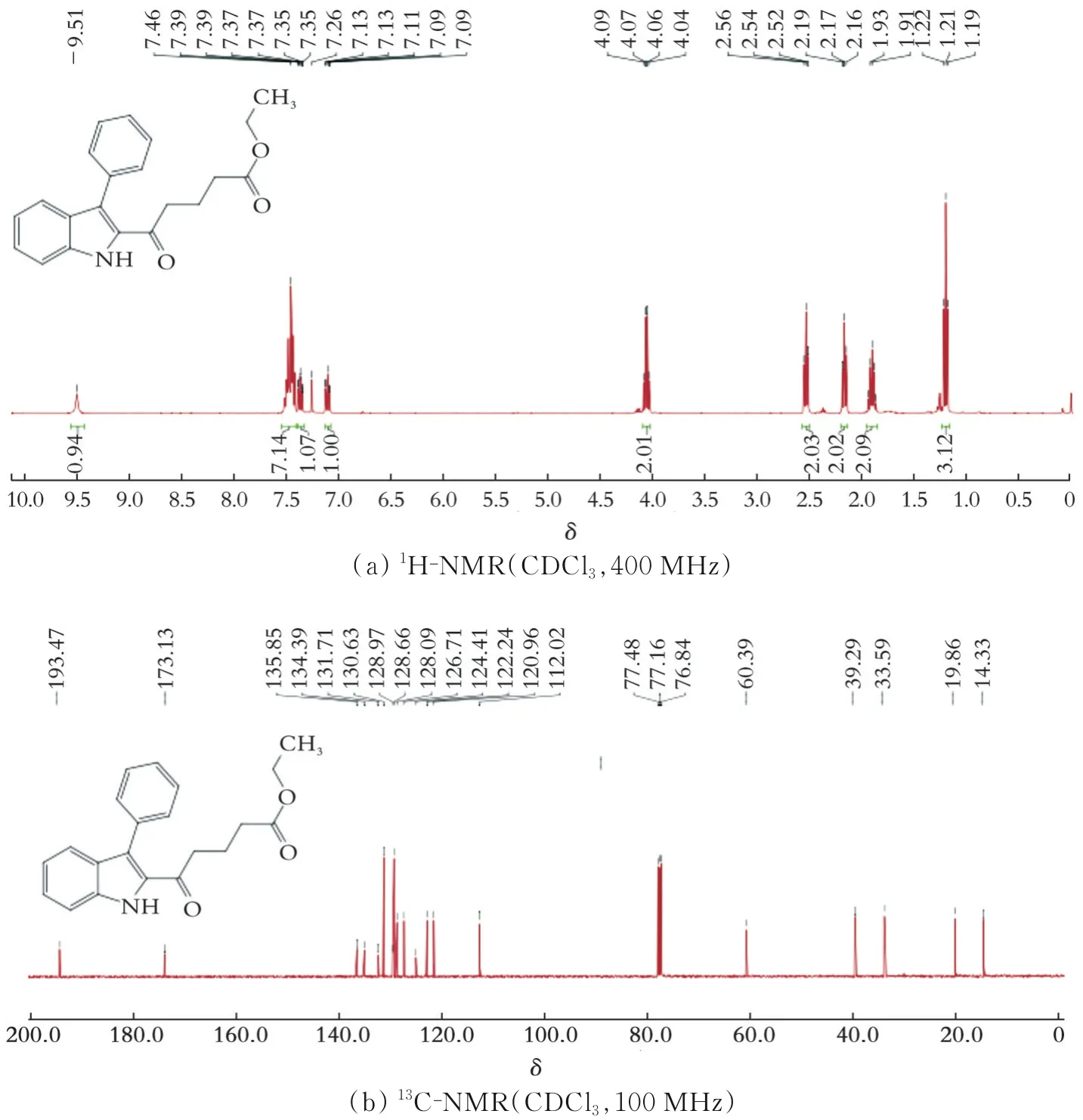
图10 化合物6的1H-NMR(CDCl3,400 MHz)及13C-NMR(CDCl3,100 MHz)
化合物7的核磁共振氢谱及核磁共振碳谱如图11所示。由图11可以看出,化合物7的核磁共振氢谱数据为:1H-NMR(400 MHz,CDCl3):δ9.58(s,1H),7.47(t,J=7.8 Hz,3H),7.39(d,J=8.3 Hz,4H),7.39(d,J=8.3 Hz,4H),7.13(t,J=7.5 Hz,1H),4.08(q,J=7.1 Hz,2H),2.55(t,J=7.1 Hz,2H),2.22(t,J=7.3 Hz,2H),2.01~1.86(m,2H),1.22(t,J=7.1 Hz,6H);化合物9碳原子的化学位移数据为:13C-NMR(100 MHz,CDCl3)δ193.15,173.14,135.83,134.19,132.90,131.97,131.71,128.96,128.75,126.84,122.80,121.91,121.19,112.15,60.49,39.38,33.50,19.69,14.33。

图11 化合物7的1H-NMR(CDCl3,400 MHz)及13C-NMR(CDCl3,100 MHz)

3 结 论
以2-位单侧链取代的1,3-环己二酮作为合成砌块,与苯基四氟硼酸重氮盐进行经典的Japp-Klingemann费舍尔吲哚反应,以高达98%的产率得到了2,3-位多取代的吲哚类似物。研究结果是对1,3-环己二酮化合物碎裂化制备吲哚化合物的创新性应用,可应用于相关吲哚生物碱的全合成研究中。
