锂离子筛研究进展
2022-05-18张理元沈如倩阳金菊
张理元,沈如倩,阳金菊,李 燕,税 亿,苏 敏
(1.内江师范学院化学化工学院,四川内江 641112;2.沱江流域特色农业资源四川省科技资源共享服务平台)
锂(Li)作为地球上最轻的金属,在许多领域,如陶瓷、润滑脂、航空航天、聚合物、金属添加剂,特别是锂离子电池中发现了各种重要应用[1]。近些年,因为可充电的锂电池在便携式的电子设备、电动器具、电动汽车和电网存储中的广泛应用,电池的锂消耗量显著增加。这些应用的激增需要利用各种可行的锂资源[2]。自然界中的锂资源主要存在于盐湖卤水、海水和矿石中。根据美国地质勘探局2018年的数据,在全球范围内持续勘探的锂资源大幅增加,到2018 年已勘探超过5 300 万t,而世界锂产量约55%来自卤水。同时,废锂离子电池(LIB)因其环境问题和经济效益而成为二次锂资源[3]。预计随着锂离子电池数量的增加,回收和再生用过的锂离子电池关注度越来越高[4-6]。世界范围内开展了大量从各种锂资源中提取锂的研究[2]。对于锂矿石,包括锂辉石、锂云母、锂磷铝石、羟硼硅钠锂和粘土,各种工艺方法,如煅烧/焙烧[7-8]、氯化[9]和酸/碱消化[10-11]得到了广泛的发展。例如,在加拿大魁北克的Nemaska锂业一直专注于通过可再生电解法将锂辉石精矿转化为氢氧化锂,西澳大利亚锂业目前正经历一段变革时期,其中,该公司计划将传统的矿产品采选转化为锂化工产品生产,主要使用SileachTM process[12]。然而,由于高温预处理、工业应用的需要和环境的限制,仍需进行大量的研究以改进和优化这些工艺,以降低运营成本和生产效率。从废锂中回收锂主要基于湿法冶金工艺,通过预处理和浸出,然后沉淀、溶剂萃取、电解或离子交换进行。然而,与原资源开采相比,由于系统的复杂性,从废锂中回收锂在经济上仍然没有吸引力[13]。目前,从盐湖卤水中提取的锂在世界碳酸锂产量中所占份额最大,因为使用传统的太阳能蒸发和沉淀工艺从卤水中提取锂仍然是最具成本效益的锂生产方法[2,14-15]。新兴的提锂技术,如溶剂萃取、离子吸附、电化学方法和膜分离等,最近也被广泛报道[16-19]。在盐水体系中,由于阳离子和阴离子的数量不同,可以形成大量的离子盐,如氯化物、硫酸盐和碳酸盐。卤水的组成决定了工艺的选择和操作难度。特别是卤水中m(Mg)/m(Li)是提锂的关键因素,这可归因于Mg2+和Li+之间几乎相同的性质。一旦m(Mg)/m(Li)超过6,从盐水中提取锂的传统沉淀法的应用就变得非常有限。然而,全球大多数已知的卤水都有很高的m(Mg)/m(Li)。在中国,超过80%的锂存在于盐水中,与南美洲相比,其m(Mg)/m(Li)通常更高(最高可达1837∶1)。为了从高m(Mg)/m(Li)的卤水中分离Mg 和Li 并回收Li,人们进行了大量的研究,在众多提锂技术中,锂离子筛吸附法因其在高镁锂比的盐湖卤水中表现出的对锂离子的高吸附选择性,加之制备工艺简单、成本低、安全环保,成为国内外研究的重点。
目前对锂离子筛的研究,主要有锰系锂离子筛[20-21]和钛系锂离子筛[22-24]。锰系锂离子筛主要通过酸处理LiMn2O4[25]、Li1.33Mn1.67O4[26]、Li1.6Mn1.6O4[27]前驱体制备,在酸洗脱锂的过程中,锰系锂离子筛前驱体中部分锰元素会由于Jahn-Teller 效应出现溶损,进而使得离子吸附剂的结构稳定性和吸附性能大大减弱,循环寿命缩短,限制了其工业化的应用。钛系锂离子筛被日本学者ONODERA 等[28]首次制备,发现其在使用过程中溶损较低、化学性质优于锰系锂离子吸附剂并对Li+具有良好的选择吸附性等优点。钛系锂离子筛[29-31]主要有层状结构的H2TiO3与尖晶石结构的H4Ti5O12两种结构,均为前驱体Li2TiO3和Li4Ti5O12经过稀酸的酸洗使氢离子替代原来晶体结构中的锂离子,然后变成H2TiO3和H4Ti5O12型吸附剂。锂离子筛吸锂/脱锂的机理在目前看来,还没有达到广泛的共识,不同的结构有不同的研究解释。本文综述了锂离子筛提取锂的机理、前驱体制备方法,对锂离子筛的应用前景进行了展望。
1 锂的插层/脱层机理
1.1 尖晶石结构
近几十年来,人们提出了氧化还原机制、离子交换机制和复合机制3种主要的机制来解释尖晶石中Li+的插层/脱层反应。
1.1.1 氧化还原机制
在HUNTER[32]的早期研究中,LiMn2O4中的锂离子脱层/插层的过程被认为是氧化还原反应。研究指出,LiMn2O4在酸性环境中的Li+脱层过程伴随着Mn(Ⅲ)的歧化,但只有表面的Mn(Ⅲ)转化为Mn(Ⅱ)和Mn(Ⅳ)。伴随着Mn(Ⅱ)的溶解,表面Mn(Ⅳ)和内部Mn(Ⅲ)开始发生转变,Li+以晶体的形式从内部逐渐扩散到表面,最终溶解在溶液之中。这一过程可以通过类似于KOZAWA 和POWERS 等[33]研究出来的碱性电极中λ-MnO2阴极还原的机制来解释,结果见图1。在Li+插层过程中,随着Li+重新进入8a四面体位置,Mn(Ⅳ)被还原为Mn(Ⅲ),这已被OOI等[34]证实。
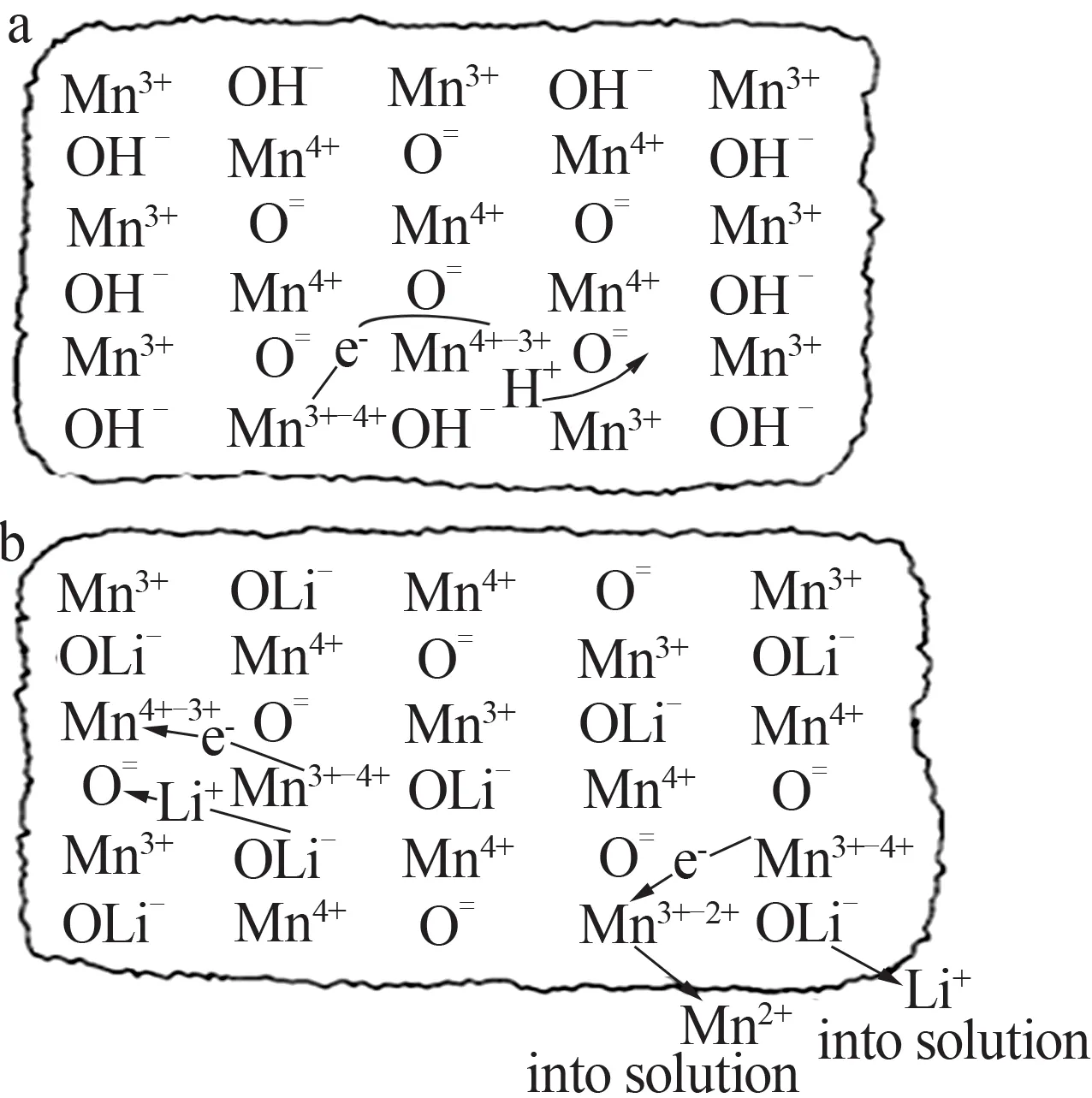
图1 阴极还原过程中λ-MnO2颗粒平衡(a);酸性水溶液中LiMn2O4向λ-MnO2转化(b)[33]Fig.1 Equilibration of λ-MnO2 particle during cathodic reduction process(a);conversion of LiMn2O4 to λ-MnO2 in acidic aqueous solution(b)[33]
Li+插层过程以表面Mn(Ⅲ)的歧化为驱动力,Mn(Ⅳ)继续保留在晶体结构中,而Mn(Ⅱ)则溶解在水溶液中。这一理论可以解释:1)尖晶石结构的锂离子筛的Li+容量在持续循环操作过程中会逐渐下降;2)由于H 原子的半径小于Li 原子的半径,Li+插层后尖晶石结构的晶格参数变小。
OOI 等[35]提出了一个阶段模型来描述Li+在λ-MnO2中的拓扑插入过程。他们指出Li+和电子迁移在λ-MnO2结构中是独立的。因为Mn(Ⅲ)和Mn(Ⅳ)的交替分布,所以电子的迁移在尖晶石结构中很容易实现。因此,Li+插入过程由以下两个步骤完成:第一,将锂离子插入λ-MnO2框架的四面体空位,同时将Mn(Ⅳ)还原为Mn(Ⅲ),通过OH-的氧化来补偿由Li+的插入而产生的λ-MnO2的电子缺陷。根据此模型可知,氧化不必靠近锂离子插入的位置,锂离子在λ-MnO2中的插入不会破坏尖晶石结构。在LiMn2O4脱锂的过程中,可以通过多种实验来验证氧化还原机理。OOI等[36]对λ-MnO2的pH滴定研究表明:在锂离子的吸附过程中消耗了等量的LiOH,这意味着从LiMn2O4中提取Li+后便生成了有空位四面体结构的λ-MnO2。在随后的研究中,根据氧化还原机理,对相同的滴定曲线进行了很好的分析。TANG 等[37]还研究发现了可用含硫的氧化剂[0.5 mol/L(NH4)2S2O8]进行该反应。氧化还原机理可以很好地解释尖晶石LiMn2O4中Li+的洗脱和吸附过程。
1.1.2 离子交换机制
在SHEN 的早期研究[38]中,温度为10~95 ℃时,可通过电解来制备锰氧化物,呈现出的离子交换行为是:在25 ℃或更低的温度条件下,氧化物具有相当于每2个锰原子1个质子的高离子交换容量;但是升高温度后,制备的产品便呈现出相应的较低容量,而在95 ℃仅表现为表面的交换。此外,加热时Li+交换固体会形成尖晶石结构的LiMn2O4,笔者认为可以通过稀酸酸洗处理将尖晶石结构的LiMn2O4转化为HMnO4。SATO 等[39]通过X 射线光电子能谱(XPS)分析表明,在Li+的洗脱和吸附过程中,表面锰离子的价态不会改变,而是发生了Li+-H+的离子交换反应,且和溶液的pH 没有关系,通过尖晶石结构的锂离子筛的傅立叶变换红外光谱分析可知表面羟基仍存在,研究发现这与尖晶石结构中的空位8a四面体的结构有关系。结果表明,洗脱Li+后,氢以羟基的形式存在。ZHANG 等[40]通过pH 滴定的实验证实MnO2离子筛具有两种交换机理:当pH≤4.46 时,四面体8a位具有较强的酸性,易于交换;当pH 高于4.46时,八面体16d位具有较弱的酸性。因此,他们认为是锂离子与质子之间发生了一种适当的离子交换反应导致了Li+在λ-MnO2中的选择性吸附。
AMMUNDSEN等[41]的中子衍射研究:Li1.33Mn1.67O4的H+交换和Li1.33Mn1.67O4的反向反应(Li+交换)表明了Li+洗脱和再吸附过程中的离子交换机理如公式(1)所示。

KIM 等[42]通过对Li1.33Mn1.67O4的四面体和八面体Li 位之间的键合性质的研究分析(如表1 和图2)表明,在这两个位置锰氧化物中的Li 都会高度电离,但是四面体位置上的锂的净电荷大于八面体位置上的锂的净电荷,因此得出四面体位置比八面体位置具有更高的Li+/H+交换性,对Li+有较好的选择性吸附作用的结论。根据这一机制,质子可以完全取代固体中的锂离子,但晶体中的Mn(Ⅲ)和Mn(Ⅳ)位在Li+-H+离子交换过程中保持不变,质子化后的固体由于保留了尖晶石结构,对Li+具有很好的可逆性和高特异性。因此,该模型解释了高浓度的OH-能促进锂离子筛(LISs)中Li+的插层的原因。

表1 晶格常数、用于计算的主要原子间距离和离子的计算值Table 1 lattice constants,major interatomic distances used for the calculations and the calculated values of ion[38]

图2 Li1.33Mn1.67O4的四面体模型(a);Li1.33Mn1.67O4的八面体模型(b)[42]Fig.2 Tetrahedral site model in Li1.33Mn1.67O4(a);octahedrel site model in Li1.33Mn1.67O4(b)[42]
近年来对锂离子在LISs 中的插层/脱层研究发现主要的插层/脱层机制是离子交换机制,但仍存在一些不完善,如它不能够用来解释有关溶液中锰的损失和在回收过程中Li+储存容量逐渐下降的观察结果。
1.1.3 复合机制
虽然从水溶液中回收锂资源的许多现象可以用氧化还原机理和离子交换机理来解释,但它们都存在各自的局限性。因此,在这两种机制的基础上,提出了复合机制。OOI等[43]研究了锂离子在尖晶石型锰氧化物中的插入反应,将锂的插入位点分成三组:氧化还原型位、锂离子特异性离子交换位和非特异性离子交换位。每个位点的比例根据准备条件的变化而变化。锰的氧化状态对热处理前驱体中插入位点的形成起着重要作用。FENG等[44]认为Li+是通过氧化还原型机制和离子交换型机制提取/插入的,锂离子的提取/插入位点可分为两种类型:氧化还原型和离子交换型。一般来说,除了晶体中Mn(Ⅲ)数量增加的情况,其余情况Li+都优先从离子交换型位点进行提取/插入。这是因为氧化还原型位点和离子交换型位点的数目分别与Mn(Ⅲ)和Mn(Ⅳ)的含量有非常紧密的关系。除此之外,他们还发现,锂锰氧化物尖晶石的制备条件(热处理温度和起始材料的Li/Mn 物质的量比)决定了这两种类型位点的比例。该复合机理相比前两种机理能更好地解释锂在水溶液中的吸附/解吸过程,但是该机理复杂,需要进一步的实验验证。
1.2 层状结构
与尖晶石结构中的锂插层/脱层相比,CHITRAKAR等[45]认为层状H2TiO3结构中的锂插层/脱层过程简单。具体地说,由于H2TiO3的交换位点很窄,只能容纳Li+和H+,因此层状H2TiO3能够在含Na+、K+、Mg2+、Ca2+等竞争性阳离子的卤水中有效地吸附锂离子;同时,层状H2TiO3可以用质子取代锂离子进行再生。前期研究中,HOSOGI等[46]用熔化的AgNO3在300 ℃处理Li2TiO3发现Ag+在(LiTi2)层不能与Li+交换,但在(Li)层可以交换形成Ag[Li1/3Ti2/3]O2层状材料。结果表明:(LiTi2)层的Li+活性低于(Li)层。因此,(Li)层中的Li+首先交换形成H[Li1/3Ti2/3]O2,然后(LiTi2)层中的Li+进一步交换形成完全交换相H[H1/3Ti2/3]O2[47]。
HE 等[48]提出了离子交换吸附模型来解释H2TiO3对锂的吸附。作者认为,H2TiO3吸附锂是通过Li+和H+之间的简单离子交换实现的,不涉及化学键的形成或断裂。如图3所示,在这个机理模型中,锂吸附后,Li+被静电吸附在HTi2层之间,而HTi2层中存在的H+并不参与锂的吸附。

图3 H2TiO3的静电吸附模型[48]Fig.3 Electrostatic adsorption model of H2TiO3[48]
MARTHI等[49]提出了基于断键和成键的锂吸附机理模型,如图4所示。由图4可知,通过热重分析、傅里叶变换红外光谱和拉曼光谱发现,HTi2层包含由Ti—O—Ti 键形成的孤立的桥接羟基,在锂吸附过程中,HTi2中孤立的O—H 基团断裂并形成新的O—Li键。氢键O—H基团存在于层间空间,为层状H2TiO3离子筛提供结构稳定性,由于其较低的酸性强度,氢键O—H 基团对锂的吸附活性低于孤立的O—H 基团。该机制涉及到表面—OH 键的断裂,揭示了锂的实际吸附容量低于理论吸附容量的原因,也解释了H2TiO3的锂吸附动力学和热力学。

图4 基于断键和成键的锂吸附机理模型[49]Fig.4 Lithium adsorption mechanism model based on bond breakage and bond formation[49]
2 锂离子筛前驱体的制备方法
如前所述,通过H+取代相应的前驱体中的Li+能得到LISs。因此,获得LISs 的实质是合成LIS 前驱体。近几十年来,人们发展了多种制备方法,包括固相合成法和软化学合成法。其中固相合成法有固相燃烧法和微波燃烧法;软化学合成法有溶胶-凝胶法、水热法、共沉淀法、熔盐合成法、喷雾干燥法和微乳液法。
2.1 固相合成法
2.1.1 固相燃烧法
固相燃烧法是制备LIS 前驱体最常用的方法。该方法首先将锂盐(通常为Li2CO3、LiOH、CH3COOLi、LiNO3)和锰盐[通常为MnO2、Mn2O3、Mn3O4、MnO、c-MnOOH、MnCO3、Mn(CH3COO)2、Mn(NO3)2]或钛盐(TiO2)以规定的化学计量比均匀混合,然后在适当温度下煅烧一定的时间。YANG 等[50]使用LiNO3和不同的锰源,包括γ-MnO2、β-MnO2、碱硬锰矿和H+型的双锰矿锰氧化物,在400 ℃时合成了结晶良好的尖晶石型Li1.33Mn1.67O4,pH 滴定结果表明,孔道尺寸相对较小的锰源制备的锂吸附剂具有更均匀的酸性中心和更少的尖晶石岩化空间位阻。ZHANG等[31]采用固相法合成了Li2TiO3,研究了原料TiO2的晶型对Li2TiO3的酸洗脱性能以及锂离子筛的吸附性能的影响。LAWAGON等[51]以Li2CO3和锐钛矿型TiO2为原料通过固相法合成Li2TiO3前驱体,系统研究了溶液pH、固液比、温度和进料浓度对H2TiO3吸附性能的影响。在不同吸附温度(从30 ℃升高到60 ℃)下确定了吉布斯自由能,在较高温度下,H2TiO3对Li+的吸附更加自发且热力学有利,吸附容量因此增加。
固体燃烧法虽然简单易行,但仍存在粉末接触不均匀,化学反应不充分,可能产生杂质和粒度分布不均匀等技术难题。为此,提出了一些改进措施。KOSOVA 等[52]采用机械化学方法制备了高度分散的尖晶石型LiMn2O4。使用球磨机混合锂和锰原料化合物,再在一定温度下焙烧。在该过程中,球磨均匀混合原料,熔融后反应物相均匀地相互扩散,不同反应物之间的接触面积增大,从而形成高比表面积的细颗粒。HUANG 等[53]首次通过室温固相配位法合成LiMn2O4-yBry纳米粒子。以醋酸锂、醋酸锰、溴化锂为原料,柠檬酸为螯合剂。将其研磨成粉末后,使用聚乙二醇混合粉末并使其反应以达到尽可能好的均匀性。然后对混合物进行程序升温煅烧,最终得到产物。与传统的固态法相比,该方法操作简单,具有较好的商业化前景。此外,它还可以达到原料混合均匀、合成温度低、粉末粒度小结果。
2.1.2 微波燃烧法
微波燃烧法是近年来发展起来的另一种固相合成方法。微波燃烧法是通过微波与反应物在分子水平上相互影响,其利用介质材料自身损耗电磁场能量通过快速运动分子的转移和转化变为热量的加热、燃烧方式。由于微波加热的原位能量转换模式,能量将在材料内部产生,而不是来自传统加热过程的外部热源。该方法可以大大提高成核速率和晶体生长速率,缩短反应时间,提高制备效率。CUI等[54]验证了流变相辅助微波合成方法在反应时间短、立方尖晶石形状规则等方面的优越性。NAKAYAMA等[55]采用微波合成技术调控LiMn2O4的晶粒尺寸。结果表明,与传统的固相反应相比,所获得的样品的晶粒尺寸较小(约0.2~0.5 μm)。CHITRAKAR 等[56]在120 ℃下、30 min 内用微波辐射制备半晶正交LiMnO2(ο-LiMnO2),最终产物为Li1.6Mn1.6O4,酸处理时Li+的洗脱率达99%,锰的溶损率小于2%。
2.2 软化学合成法
软化学合成法旨在改善固相合成法中非均相混合物的缺点。在这些方法中,通过将可溶锂化合物和锰或钛化合物溶解到水溶液中来制备原子级的起始材料混合物。在特定的环境下,对均匀的混合物进行焙烧,可以得到最终产品。在此,介绍一些常用的软化学合成法制备LIS前驱体。
2.2.1 溶胶-凝胶法
众所周知,溶胶-凝胶法通常用于制备化学均匀性好、纯度高、相分布均匀的纳米粒子[57-59]。该工艺是将原料(可溶锂源和锰/钛源)分散在溶胶中,然后团聚形成具有连续三维网络的凝胶。再在适当的条件下加热凝胶,得到产物LIS 前驱体。在此过程中,起始材料在多组分体系中以原子尺度混合,从而使LIS前驱体完美结晶。
SUN等[60]以含有聚丙烯酸(PAA)的金属乙酸酯水溶液为螯合剂合成了尖晶石LiMn2O4粉末。结果表明,溶胶-凝胶法相比于传统的固相反应,具有低的煅烧温度和煅烧时间,这主要是由于均匀凝胶制备的混合材料具有较高的烧结性。并且,还发现可以通过改变热解的条件和螯合剂的用量来控制LiMn2O4的粒径、比表面积和微晶形貌等物理化学性质。
此外,还有很多关于溶胶-凝胶法的制备条件和机理的研究分析。SEYEDAHMADIAN 等[61]探究了溶胶-凝胶法制备纯尖晶石型LiMn2O4的最佳条件:1)pH 控制在4~6;2)柠檬酸与金属离子的物质的量比为1;3)以硝酸锂和硝酸锰为金属离子源;4)以乙醇为溶剂。该研究为溶胶-凝胶法制备纯尖晶石LiMn2O4提供了重要的指导意义。KUSHIDA等[62]研究了溶胶-凝胶法合成尖晶石LiMn2O4的结晶机理,发现尖晶石LiMn2O4的结晶过程始于280 ℃,这归结于Li-Mn凝胶中释放出的C和H原子的燃烧以及随后发生的晶化过程,在晶化过程中Li、Mn 和O 原子发生重排,结果表明,Li-Mn 凝胶前驱体的最低退火温度应在290 ℃以上。
2.2.2 水热法
水热法是指在密封的压力容器中,以水为溶剂,在高温高压的条件下进行的化学反应。水热法制备LIS前驱体克服了生产原料不均匀、操作过程复杂、设施设备成本高等缺点。此外,由于具有独特的均相成核的机理,可以创造出新的化合物[63-64]。例如,CHITRAKAR 等[56]通过加热LiMnO2,在400 ℃下,通过水热法获得相应的LIS 材料(MnO2·0.5H2O),首次合成了Li1.6Mn1.6O4,理论上产生最大锂吸收值(72.29 mg/g)。而采用固相法则无法制备,因为在煅烧n(Li)/n(Mn)为1 的金属源混合物时,会生产MnO2、LiMn2O3和Li2O 等杂质。水热法还有一个优点是:可通过改变水热条件制备不同形貌的LIS 前驱体,如纳米棒、纳米线、立方形状和球形等。而众所周知,具有特定形态的纳米吸附剂对吸附行为的影响是多种多样的[59-68]。对于LIS 晶体不同的形貌可能会影响其吸锂能力、Li+吸附平衡时间、锂的选择性和重现性。此外,LIS 晶体结构中的Li+洗脱/吸附过程将更加容易。
2.2.3 共沉淀法
共沉淀法是指在溶液中有两种或多种阳离子均相存在于溶液中,加入沉淀剂经沉淀反应后,可得到各种成分的均一的沉淀,是制备含有两种或两种以上金属元素的复合氧化物超细粉体的重要方法。共沉淀法是一种被广泛应用的制备掺杂型锂离子筛前驱体的软化学方法。FENG 等[69]以LiOH、Mg(NO3)2和Mn(NO3)2为金属源制备LiMg0.5Mn1.5O4。酸处理样品对碱金属离子中的Li+具有较高的选择性和较大的容量。然而,共沉淀法制备的掺铝和掺铁LiMn2O4样品(LiAlMnO4和LiFeMnO4)的锂容量比掺镁样品的锂容量差[70]。这可能是由于Mg 的酸洗脱率(约50%)高于Al 和Fe(均小于10%)。为了研究四面体取代产物对锂容量的影响,LIU 等[71]采用改进的草酸盐共沉淀法,通过控制前驱体溶液的pH,在前驱体中引入过量的Li 源,合成了尖晶石LiNi0.5Mn1.5O4。他们发现前驱体溶液的pH 可以影响目标材料的形貌、化学计量和晶体结构。NAGHASH等[72]通过共沉淀法制备了尖晶石LiMn2O4细颗粒。该过程先将LiNO3和MnSO4溶解在溶液中,然后在氮气气氛下,将溶液逐滴添加到四甲基氢氧化铵(TMAH)中并搅拌。用醋酸将溶液的pH 调节到8~10 后,在过滤前将形成的沉淀陈化1 h。最后,以1 ℃/min的升温速率来煅烧沉淀,得到LiMn2O4。共沉淀法具有操作步骤简单、成本低、实验条件容易控制等优点,但沉淀剂的加入会导致局部的离子浓度过高,从而导致粒径分布不均匀。
2.2.4 其他方法
制备LIS前驱体的方法很多,但应用范围有限。ZHANG 等[73]采用无机沉淀-胶溶法合成了层状H2TiO3的前驱体Li2TiO3。HELAN 等[74]采用熔盐法,在900 ℃下使用氯化锂和硫酸锰的共晶混合物制备LiMn2O4粉末,终产物具有良好的结晶度。还有研究用LiCl-Li2CO3-Mn(CH3COO)2·4H2O 制备LiMn2O4以及用LiCl-MnO2-SmCl3·6H2O 制备LiSm0.01Mn1.99O4[75-76]。SINHA 等[77]采用微乳液法制备了亚微米级的LiMn2O4尖晶石颗粒。经900 ℃退火,得到的粉体为纯晶尖晶石结构。KIM 等[78]以LiNO3、Li(CH3COO)·2H2O 和Mn(CH3COO)2·4H2O 为原料,采用溶液燃烧法合成Li4Mn5O12。WU等[79]通过烧结前驱体合成了尖晶石型的LiMn2O4,该前驱体由对应金属的醋酸盐溶液在600 ℃喷雾条件下干燥15 h后制得,所得样品颗粒细小、分布窄、结晶良好。
3 锂离子筛应用
3.1 卤水中锂的回收
卤水是锂回收的重要潜在资源之一。从经济和科学的角度来看,以下几点对于从卤水中回收锂很重要:1)卤水的可用性和太阳蒸发的适宜性;2)卤水中锂的浓度;3)碱土和碱金属与锂的比率;4)化学相的复杂性。含锂卤水资源可分为蒸发卤水、地热卤水和油田卤水3 种类型。在卤水蒸发过程中,约50%的初始天然卤水中仍有锂残留。这种现象是由于沉淀盐对锂的保留造成的。与Na+和K+相比,剩余卤水中含有大量的Mg2+,这使得从剩余卤水中回收锂变得更加困难[80]。锂回收的典型生产技术有溶剂萃取、沉淀和浮选等多种技术。CHAGNES等[81]提出了从盐水和海水中生产锂的一般流程图。在提议的工艺中,离子交换、液-液萃取、吸附和电渗析是生产锂产品前浓缩锂所需的重要湿法冶金工艺。
PELLY 等[82]报道了从死海卤水和终端卤水(钾盐生产后)中回收锂作为铝酸锂沉淀的方法,要达到90%的提取效率,需要通过稀释来控制盐水的pH,终端盐水的最适pH 应在6.8~7.0,死海盐水的最适pH 应在6.6~7.2,最佳反应时间为室温下3 h;SUN等[83]采用反应耦合分离技术从高m(Mg)/m(Li)和高m(Na)/m(Li)的卤水中分离出Li,实现了锂的高效提取,为锂的可持续提取和锂基产品的开发提供了广阔的前景。
3.2 海水中锂的回收
近十年来通过离子交换和吸附法从海水中回收锂对研究人员的吸引力越来越大。近年来,利用不同类型的膜回收锂的研究已经有很多。膜法回收锂工艺相当先进,引起了世界各国研究者的重视。有团队采用静电纺丝、热退火等方法制备了聚砜(PSf)基混合基质纳米纤维,用Li0.67H0.96Mn1.58O4或MO酸浸活化锂离子筛,并将混合基质纳米纤维与颗粒锂离子筛分散在一起,作为一种流动膜Li+吸收剂,PSf基体有效地提高了Li+的选择性。混合基质纳米纤维膜在最小的跨膜压力下具有很高的透水性。通过保持混合基质纳米纤维的动态Li+吸附能力,以及连续的流动操作,成功地控制了吸附-解吸循环时间(24 h)。通过在小体积的酸溶液中重复Li+解吸,成功地实现了Li+的富集。LIMJUCO 等[84]为了实现合成H2TiO3(HTO)在锂离子连续回收中的潜在用途,先采用固相法合成HTO,然后制备了以亲水聚乙烯醇(PVA)为基体的HTO/PVA 复合泡沫。通过化学交联,使得HTO均匀负载到泡沫表面。亲水泡沫孔隙间的连通促进了H2TiO3对Li+的高吸附性。此外,将该复合材料应用于海水中吸附Li+时表现出高选择性,经过多次使用后,能保持其吸附性能和结构完整性,表明其非常适合长期应用于从海水等液体资源中开采锂。
4 结论与展望
随着锂电池的发展,未来社会对锂资源的需求不断增大,从储量大、品位低的盐湖卤水、海水等液态锂资源中提取锂是未来的趋势。锂离子筛型吸附剂因为具有较高选择吸附性一直都是研究的热点。与锰锂离子筛相比,钛锂离子筛具有更高的结构稳定性,是近年来研究的重点。钛锂离子筛的未来发展趋势是:1)进一步增强结构的稳定性。在洗脱过程中,前驱体的转型是分步进行的,主要表现为(-133)(-206)(062)晶面衍射峰很快消失,而(-131)(002)晶面衍射峰接下来消失,紧接着出现晶型转变,出现锐钛型衍射峰,并且随着晶型转变的出现,离子筛的吸附容量下降,这也是钛锂离子筛在循环使用过程中性能降低的主要原因,通过掺杂等方式能够在一定程度上增强离子筛骨架的稳定性,抑制骨架的晶型转变。2)将锂离子筛负载在一些合适的基体上。目前采用各种方法制备的锂离子筛基本都为粉体状,由于盐湖卤水、海水中锂含量较低,采用粉体锂离子筛直接作为交换吸附剂,为达到操作交换容量,会存在反复固液分离(过滤阻力很大)、洗涤、淋洗、再生、富集困难及生产效率低等问题,难以实现工程化。将锂离子筛负载于适宜基体上是保证其洗脱、吸附性能和回收的有效途径。基体对离子筛性能影响很大,因为适宜的基体能改善离子筛的物理性质,增加有效表面和提供合适的孔结构,增强离子筛抵抗外界破坏及侵蚀的能力。另外,将锂离子筛负载于适宜基体上有助于实现离子交换柱持续、循环操作。为锂离子筛的工程化应用奠定坚实的基础。