Peutz-Jeghers综合征双胞胎患儿病例报道并文献复习
2022-05-17于敬红于淑凤宋振凤陈云庆王彩霞
于敬红,于淑凤,宋振凤,赵 燕,陈云庆,王彩霞△
(青岛大学附属医院:1.儿童医学中心;2.病理科,山东 青岛 266555)
Peutz-Jeghers(P-J)综合征是一种常染色体显性遗传病,由STK11(LKB1)基因突变引起,主要临床表现为皮肤黏膜色素沉着和胃肠道多发错构瘤。本文报道的1例先证者为双胞胎次子,因便血及腹疼就诊,查体可见典型皮肤色素沉着,胃肠镜检查发现典型息肉,外周血及口腔黏膜细胞全外显子基因检测提示STK11基因突变。先证者胞兄临床表现及胃肠镜检查较先证者轻,STK11基因未见异常,其父母无P-J综合征相关临床表现,外周血和口腔黏膜细胞基因检测未见STK11突变,内镜检查亦未见特征性息肉。本例先证者突变基因及基因突变方式较少见,现报道如下。
1 临床资料
先证者,男,12岁,为双胞胎次子,因“间断便血15 d伴腹痛2 d”于2020年10月31日入当地医院。15 d前先证者无明显诱因出现大便表面带血,为暗红色,伴有肛门肿物脱出,无恶心、呕吐,无腹胀、腹泻,无排便困难及疼痛,肿物于当地医院回纳,未再脱出,大便亦未再带血。2 d前先证者出现阵发性左上腹胀痛,与进食无明显关系,持续1~2 min后可自行缓解,无胸闷、憋气,无心慌、气短,无恶心、呕吐,无便血。询问病史得知先证者出生后口唇即有少许黑色扁平斑点,不凸出于口唇黏膜表面。随年龄增长斑点渐增多,部分融合成片,且手指末端出现黑斑,颜色逐渐加深,未予诊治。入院后查体:先证者神志清,精神好,口唇黑斑,部分融合成片,未突出皮面,口腔黏膜可见1处黑斑,手指散在黑斑(图1),腹部柔软,无包块,左下腹压痛,心肺查体未见明显异常。
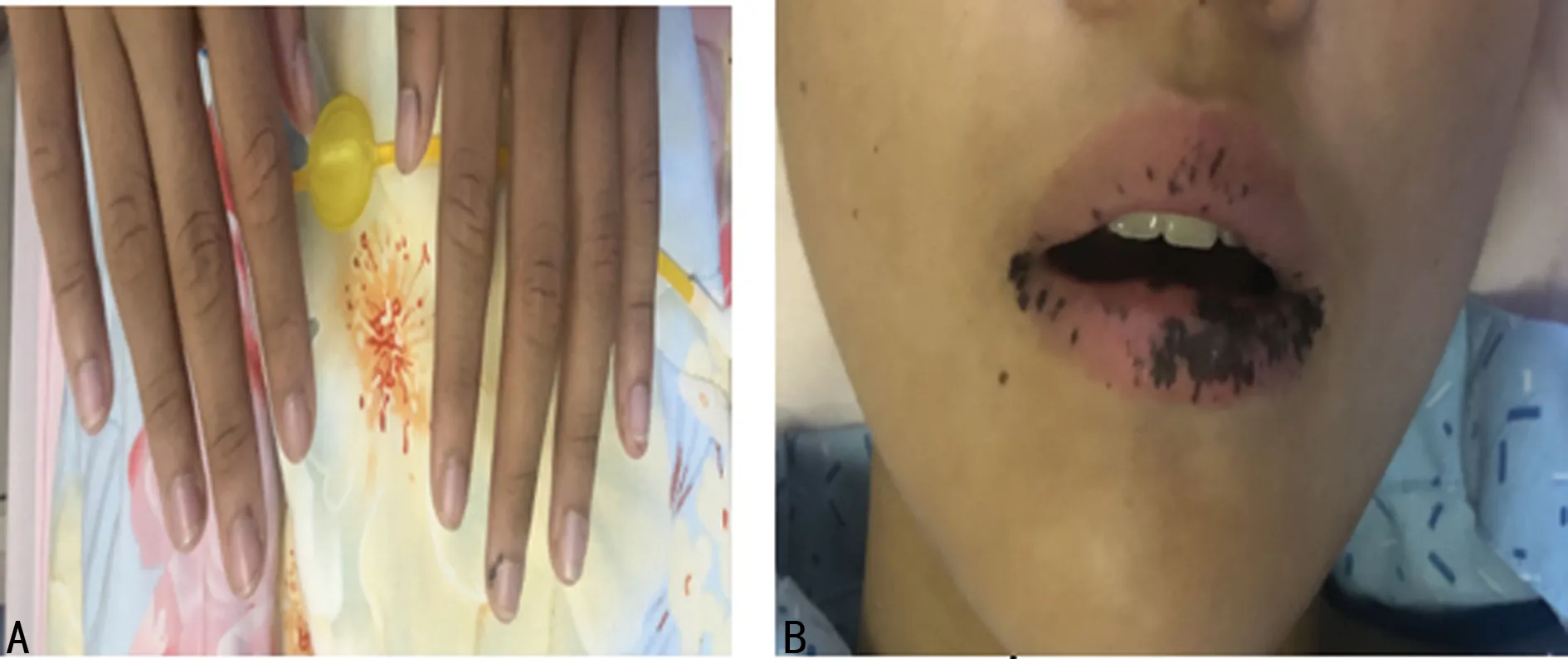
A.手指黑斑;B.口唇黑斑。
先证者入院完善相关检查,排除禁忌证后行结肠镜及胃镜检查。内镜下可见胃及肠内多发息肉,直径2~20 mm,形状以半球型为主,部分呈结节分叶状(图2A、2B)。病理提示:胃增生性息肉并管状腺瘤,十二指肠降段增生性息肉,肠黏膜组织呈息肉样增生。为进一步明确小肠内息肉增生情况,行小肠CT造影,结果示小肠多发息肉(图2C)。治疗过程中,先证者出现肠套叠(小肠-小肠型,图2D),及时予腹腔镜手术切除套叠肠管,并送病理。免疫组织化学检查示:CK7阴性,CK20阳性,MUC5AC阴性,MUC2部分阳性,结蛋白阳性,平滑肌肌动蛋白阳性,D2-40阴性,Ki-67部分阳性。在先证者监护人均知情同意并经过医院伦理委员会(批件号:QYFYWZLL26508)批准下,完善外周血及口腔黏膜细胞全外显子基因检测,提示基因STK11发生c.291-1G>A突变(chr19:1218416-1218416),且为低比例嵌合,导致剪接位点发生改变,与常染色体显性遗传病P-J综合征相关。因先证者确诊P-J综合征,而先证者胞兄具有相同的口唇及十指黑斑,为明确诊断入本院。先证者胞兄无明显不适,查体可见口唇及十指散在黑色斑点,未突出皮面,无融合,较先证者轻。行小肠CT造影示部分性中肠旋转不良,排除禁忌证后完善胃肠镜检查。内镜下可见散在大小不等息肉,直径可达10 mm,形状以半球形为主。胃部病理示腺体性息肉,肠道病理示增生性息肉。对先证者胞兄外周血及口腔黏膜细胞行全外显子基因检测均为野生型,未见STK11基因突变。见图3。

A.胃镜下部分息肉表现;B.肠镜下部分息肉表现;C.息肉病理表现(200×);D.先证者腹腔镜下肠套叠。

A.先证者外周血基因检测;B.先证者父亲外周血基因检测;C.先证者母亲外周血基因检测;E.先证者口腔拭子基因检测;F.先证者胞兄外周血基因检测;G.先证者胞兄口腔拭子基因检测。
对先证者3代亲属进行调查,未见与先证者具有相同或相似临床表现的家属,未发现因消化道息肉行相关手术或有相关恶性肿瘤家属。同时对先证者父母进行外周血全外显子基因检测,未发现STK11基因突变。先证者胞兄临床表现轻,内镜下行胃肠道息肉切除后未见明显不适,大便颜色、形状正常。先证者因小肠套叠行腹腔镜部分肠管切除术,术后恢复好,未见明显不适。出院后定期随访双胞胎患儿恢复情况,目前双胞胎患儿病情稳定,无不适。
2 讨 论
P-J综合征以皮肤黏膜色素沉着和胃肠道多发性错构瘤息肉为主要临床表现,其发生率为1/8 600~1/25 000[1]。皮肤黏膜色素沉着主要位于口唇、肛周、手指或足趾,1岁以后颜色加深,除口腔黏膜外,其他部位色素沉着均在青春期以后逐渐消失[2]。息肉主要位于胃肠道,近几年也有鼻、胆囊、输尿管等消化道以外息肉的报道[3-4]。P-J综合征患者特征性临床表现出现时间不一,有的出生后即存在,有的则在成年后出现。国外对P-J综合征特征性临床表现出现的中位时间进行研究时发现,该病出现的时间与先证者为家族遗传还是散发病例无关[5]。儿童时期的主要临床表现为息肉导致肠套叠、贫血、梗阻等,成人时期则面临各系统患癌风险的增加[6]。本例先证者即因肛门肿物脱出就诊而发现。
P-J综合征为临床罕见病,关于指导该病管理的临床资料和科学数据较少见。随着基因工程的不断发展,医学界对P-J综合征认识不断深入,其临床诊断标准不断更新。1997年,P-J综合征临床诊断标准简单描述为:(1)胃肠道典型息肉或胃肠道有1个息肉;(2)伴有P-J综合征典型色素沉着或P-J综合征家族史。2010年更新为:有2个或以上组织学典型P-J综合征息肉;(2)或近亲中P-J综合征家族史个体有任何数量的典型息肉;(3)或近亲中P-J综合征家族史个体有特征性黏膜皮肤色素沉着及任何数量的P-J综合征息肉[7]。当然确切诊断还需行基因检测,明确有STK11基因突变。P-J综合征息肉的形成主要与转化生长因子β(TGF-β)相关。LKB1基因突变导致TGF-β不能正常表达,使其失去调节控制上皮细胞增殖作用,从而形成息肉。关于息肉治疗,欧洲消化道内窥镜协会建议,对于大于8岁的无症状P-J综合征患者,应每1~3年完善1次胃肠道全面检查,若小肠典型息肉直径为>15~20 mm时应进行选择性息肉切除术,以防止肠套叠[4]。国内有关P-J综合征患者的研究显示,患者黏膜色素斑极少发生恶变。研究者在追踪多例P-J综合征患者疾病转归情况时发现,患者胃肠道肿瘤发生率较普通人明显升高,且乳腺、膀胱、食管等肠外肿瘤发生率也会增加。
STK11基因位于常染色体19p13.3,共含有10个外显子,其中外显子1~9为基因编码区,10为非翻译区。有研究利用直接测序或多重连接扩增技术发现,P-J综合征患者常见的基因突变主要发生在外显子1~9[8]。2018年关于P-J综合征基因突变种类已报道400余种[9],主要为点突变和缺失突变,低比例嵌合相对少见,发生在内含子的低比例嵌合更为少见。STK11基因编码丝氨酸/苏氨酸激酶,参与控制细胞周期停滞、p53介导的细胞凋亡、细胞外因子和TGF-β信号传导、ras基因诱导的转化、能量代谢等[10]。因此,P-J综合征患者癌变概率增加的主要原因为STK11基因突变,其使抑癌基因P53表达降低,由P53介导的细胞增殖和凋亡失去调控,从而导致异常细胞增殖[11]。STK11基因的氨基端为非催化域,羧基末端为调节域,可使丝氨酸及苏氨酸被磷酸化,从而发挥磷酸化和RNA翻译后修饰的作用[5,9]。P-J综合征双胞胎患儿相对少见,目前相关研究显示,双胞胎患儿无论临床表现还是基因突变位点、类型均相同[12]。本例先证者外周血及口腔黏膜细胞STK11呈内含子c.291-1G>A的低比例嵌合状态,且双胞胎患儿基因不同。
低比例嵌合体指个体体内同时存在两套基因:一种为正常基因,占绝大部分;另一种为突变基因,所占比例相对较少。当突变基因占据一定比例后可在遗传表达中发挥作用,影响个体临床表型。目前,国内外关于P-J综合征低比例嵌合报道不多见,相关报道也只是粗略描述了患者无相关临床表现,均因具有P-J综合征临床表现的亲属确诊而确诊[13]。本例先证者虽是低比例嵌合,但具有P-J综合征特异性临床表现,且先证者胞兄具有相关临床表现及胃肠道息肉,但STK11基因却未见突变。相关研究显示,P-J综合征存在明显的遗传异质性和表型异质性,一些患者只表现出皮肤黏膜色素沉着,而一些患者既有皮肤黏膜色素沉着,又有胃肠道多发息肉,甚至一些患者临床表现已经达到P-J综合征诊断标准,但其STK11基因却未检测到突变[14-15]。本研究中,先证者胞兄体现出遗传与表型异质性特点,推测该例患儿基因嵌合比例更低或存在目前检测手段尚不能检测出的突变。各种突变致使DNA在转录过程中出现错译或无义突变等导致转录提前终止,最终致使RNA翻译的蛋白质链变短或蛋白质结构发生改变而失去相应功能。随着基因工程的不断发展,在P-J综合征患者中发现,一些内含子突变同样致病。其原因为距离外显子两端较近的内含子部分碱基序列组成剪接位点,当内含子与外显子同时被转录在一条原始RNA中时,原始RNA通过剪接位点剪切掉内含子片段,而外显子根据剪切位点会重新组合为成熟mRNA。因此,当外显子两端的内含子碱基发生变化时可直接导致剪接位点发生变化,进而导致mRNA碱基序列或碱基数量改变,使翻译出的蛋白质链长度或结构发生改变,从而致病。
综上所述,P-J综合征是一种临床罕见病,具有较低发生率,其发病机制及遗传特点目前尚未阐明。P-J综合征患者成年后各系统癌症发生率较高,因此一旦确诊应及时切除息肉,并定期进行内镜检查,以改善预后。本研究中,具有P-J综合征典型临床表现的同卵双胞胎患儿基因突变存在差异,双胞胎长子未检测出基因突变,而双胞胎次子基因突变方式为STK11基因内含子低比例嵌合,其父母无P-J综合征临床表现,也未检测出相关基因突变。结合本病例资料,作者推测P-J综合征可能存在其他基因突变或目前检测手段难以检测出的更低低比列嵌合等致病原因。因此,建议对于有疑似P-J综合征临床表现的患儿,除进行胃肠镜、组织病理及血液基因检测外,可增加病理组织的基因测定,从而为P-J综合征的研究及指导优生优育提供可靠资料。
