儿童Alport综合征合并GJB2基因突变1例分析并文献复习
2022-05-16成艳辉黄惠梅
成艳辉,包 瑛,骞 佩,黄惠梅
(1.西安医学院,陕西 西安 710021;2.西安交通大学附属儿童医院(西安市儿童医院) 肾脏科,陕西 西安 710003)
Alport综合征(Alport syndrome, AS)是一种遗传性肾脏疾病,由编码Ⅳ型胶原的α3、4、5链的COL4A3、COL4A4、COL4A5基因突变引起[1]。典型临床表现为血尿伴或不伴蛋白尿、进行性肾功能减退、感音神经性耳聋及眼部异常[2]。AS有3种遗传方式,最常见的为X连锁显性遗传型AS(X-linked Alport syndrome, XLAS),约占85%; 其次为常染色体隐形遗传型AS(autosomal recessive inheritance, ARAS)和常染色体显性遗传型AS(autosomal dominant inheritance, ADAS),约占15%和5%[3-4]。AS是导致终末期肾病(ESRD)的主要原因之一,目前尚无根治方法。GJB2是第1位导致耳聋的致病基因,约50%的遗传性耳聋由GJB2引起,其变异可能导致常染色体显性遗传或隐性非综合征性听力损失以及综合征性听力损失[5]。GJB2基因突变者听力学表型多样, 主要表现为语前、双侧对称性的,中度至极重度感音神经性聋, 此外也可表现为单耳耳聋、双耳轻度感音神经性聋及迟发性、渐进性耳聋[6-7]。
1 临床资料
患者,女,3岁8个月,因“发现肉眼血尿8天”于2020年9月26日入院。病初患儿无明显诱因出现全程肉眼血尿2~3次,家属未在意,血尿自行好转。后因“发热、咳嗽”再次出现肉眼血尿,就诊于当地医院,予抗感染对症治疗1周后,仍有间断血尿,遂就诊于西安市儿童医院肾脏科。病程中患儿无浮肿、少尿,无高血压,无关节肿痛,无皮疹,无脱发及口腔溃疡。入院体格检查:体温36.5 ℃,心率118次/min,呼吸24次/min,血压108/70 mmHg(1 mmHg=0.133 kPa);颜面部、双下肢无浮肿;双肾区叩痛阴性;余查体无异常。实验室检查:尿常规潜血4+,蛋白+,白细胞+。血超敏C反应蛋白0.5 mg/L,血常规:白细胞计数12.42×109/L,中性粒细胞计数8.46×109/L,血红蛋白118 g/L,血小板计数406×109/L;24小时尿蛋白定量179.69 mg/kg;肝肾功、心肌酶、电解质、补体均正常,自身抗体均阴性;乙肝定性、HIV、梅毒、丙肝均为阴性;胸片未见明显异常征象。泌尿系彩色超声正常。心电图正常。听力检查:听性脑干反应(auditory brainstem response, ABR)检查结果:双耳70 dB nHL给声,Chrip声刺激,均可见Ⅰ、Ⅲ、Ⅴ波反应,各波波形分化好,且各波潜伏期均未见延长,双耳Ⅰ-Ⅲ、Ⅲ-Ⅴ、Ⅰ-Ⅴ波间期正常。Ⅴ波反应阈:右耳30 dB nHL,左耳50 dB nHL。骨导ABR检查结果:双耳40 dB nHL给声,Chrip声刺激,Ⅴ波潜伏期均未见延长。Ⅴ波反应阈:右耳20 dB nHL,左耳30 dB nHL,双耳750~8 000 kHz, 均未记录到DP声强。双耳耳纤维内镜:分泌性中耳炎(左),声导抗示左耳C型曲线。肾脏穿刺活检病理,免疫荧光:6个肾小球。IgG-, IgM++, IgA-,C3+, C1Q-, FRA-。光镜:镜下可见2条肾皮质,15个肾小球,系膜细胞和基质轻度增生,节段性内皮细胞增生,基底膜无明显异常;肾小管上皮细胞空泡及颗粒变性,可见红细胞管型;肾间质及小动脉无明显病变。结合免疫荧光检查及临床,符合轻度系膜增生性肾小球肾炎。电镜:肾小球毛细血管基底膜弥漫菲薄,基底膜厚度100~170 nm,未见电子致密物,上皮细胞足突节段融合。肾小管、肾间质无特殊病变;符合薄基底膜肾病,见图1。
为进一步明确诊断,分别抽取患儿及其父母2 ml外周血送至北京智因东方转化医学研究中心,进行全外显子组测序和生物信息学分析,检测到COL4A5基因 c.555(exon10)-c.568(exon10)delTGGTCCCACTGGTA突变,GJB2基因c.109(exon2)G>A突变, 见图2~3。根据美国医学遗传学与基因组学学会(ACMG)指南(2015)及遗传学模式分析患儿基因结果:①c.555(exon10)-c.568(exon10)delTGGTCCCACTGGTA突变的证据强度为“PVS1+PM2+PM6”,判断为致病性突变。先证者杂合新发(denovo)突变,符合X染色体显性遗传(XD)疾病发病机制,先证者及其家系成员表型及基因型的共分离:符合。变异支持致病。②c.109(exon2)G>A证据强度为“PS4+PP1-Strong+PM3“,判断为致病性突变。先证者为纯合子,符合常染色体隐性遗传(AR)疾病发病机制,先证者及其家系成员表型及基因型的共分离:符合。变异支持致病。

图1 患儿肾脏组织病理 a.HE染色;b. 透射电子显微镜照片
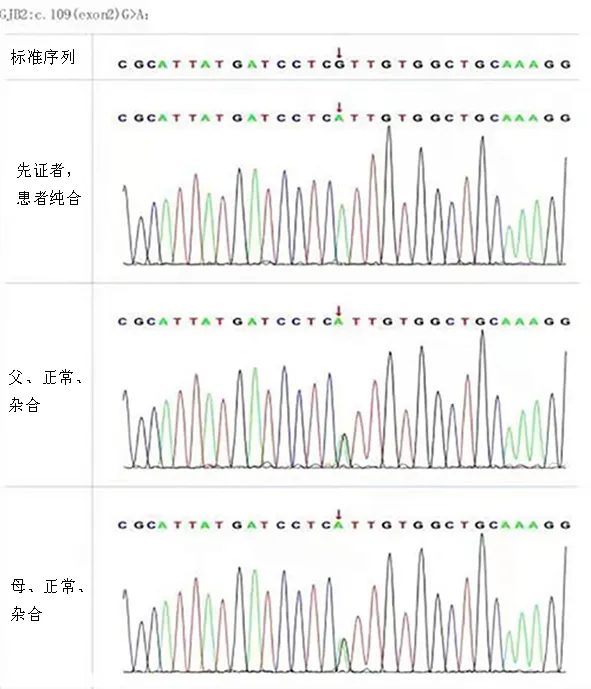
图2 患儿及父母GJB2基因测序图

图3 患儿及父母COL4A5基因测序图
2 讨 论
AS是常见的遗传性肾炎之一,由编码Ⅳ型胶原蛋白的基因突变导致。Ⅳ型胶原蛋白是肾脏、耳蜗和眼睛基底膜的重要组成部分。由6种不同的α链组装成3种不同的异质三聚体[8]。α3,α4和α5(Ⅳ)链的缺陷会形成异常的成熟α链,进而破坏Ⅳ型胶原分子的形成,导致基底膜结构的改变。 这些生理变化最终导致肾小球、视网膜和耳蜗等器官的功能障碍[9]。
听力损伤是AS的主要表现,也是早期症状之一。AS听力损伤的发生率为50%~67%。由于 AS导致的听力损伤不是先天性的,患者的新生儿听力筛查往往是正常的,通常在儿童晚期或青春期早期首次通过听力测量法检测到,表现为进行性双侧对称中高频的听力损伤[3]。
不同人群听力损伤的发病率不同,不同遗传方式导致的听力损伤亦不同。由于Ⅳ型胶原分子各α链表达具有组织差异性,在耳蜗组织中α3~α6链均有表达,但由于α5链是两种Ⅳ型胶原蛋白分子的主要组成成分,所以XLAS患儿更易出现听力损伤[10]。 有研究表明,XLAS男性患儿听力损伤发生率约为66.27%,而女性患儿仅为20.31%,且女性患儿的听力损伤多为轻度,男性往往症状较重[10-11]。除此之外,差异还体现在发病年龄上。男性听力损伤多发生在学龄期儿童,但女性多为中年以后。这与先前Jais等[12-13]报道一致。XLAS的听力损伤程度在种族方面亦有差异,国外相关文献报道的语言频率范围听阈平均值为50 dB, 明显低于张晓等[10]报道。 听力损伤还与基因突变类型有一定联系。 XLAS男性患者听力损伤表现与基因突变的严重程度有着明显的相关性[14]。在30岁之前发生听力损伤的错义突变患者约为60%,而其他类型的突变共达到90%[15]。女性则无明显相关性。此外,听力损伤与肾功能损伤有较强的相关性,是ESRD的一个预测因子。殷佳珍等[16]研究发现听力异常组AS患儿比听力正常组的肾功能预后显著较差。本例为XLAS女性患儿,以血尿、蛋白尿起病,听力检测左耳轻度感音神经性耳聋,基因检测提示COL4A5基因 c.555(exon10)-c.568(exon10)delTGGTCCCACTGGTA突变。但患儿出现感音神经性耳聋的年龄相较文献报道早,可能与其合并GJB2基因突变相关。
GJB2基因是首个被发现的耳聋致病基因,也是国内最常见的耳聋致病基因,约占50%以上[17]。据报道,正常人GJB2基因的携带率为2%~3%[18]。该基因位于人类染色体13q11~12,主要编码Cx26蛋白,其与相邻的缝隙连接蛋白组成了细胞间信息传递的通道。当GJB2基因突变时,会导致内耳感觉细胞和支持细胞间的离子和信息传递障碍[19]。
GJB2基因突变临床表型多样,主要导致以语前、双侧对称性为特征的先天性非综合征性耳聋,听力损伤为中重度到极重度[20]。此外,由于GJB2基因突变位点多种多样,存在一定的外显率,并且基因型与听力受损外显时间存在一定的关系,GJB2的突变可表现为出生后迟发性、渐进性耳聋[21]。不同人种GJB2常见的基因突变形式存在明显的差异性。研究表明,欧洲地区以c.35delG位点突变最为常见;犹太地区以c.167del突变最常见;亚洲地区以c.235delC位点突变最为常见[7, 22]。
2011年纪育斌等[23]对中国非综合征性耳聋患者进行GJB2基因突变流行病学研究发现,国内常见4种热点突变c.235delC、c.299delAT、c.176delI、c.35delG,其检出率分别为11.9%,2.22%、0.65%、0.27%。近期研究表明,我国c.109G>A(p.V37I)检出率也较高,约为6.7%。大量研究证明GJB2基因p.V37I突变的听力表型多样,可为轻至重度听力损失[7, 22-25],但主要表现为轻至中度感音神经性听力损失[25]。本例患儿基因检测出c.109G>A(p.V37I)突变,新生儿期听力筛查通过,学龄前期听力检测提示左耳轻度感音神经性耳聋,与此前报道相符。
目前尚未发现AS合并GJB2基因突变的报道,对于COL4A5基因合并GJB2基因突变听力损伤的临床预后尚不清楚。但是张晓等[10]通过对49例AS患儿随访2年发现, 28例患儿听力水平呈下降趋势,每年约5 dB nHL。Chan等[26]通过对18例GJB2基因c.109G>A纯合突变或复合杂合突变的患儿研究发现,其中有7例(39%)出现听力进展,平均进展8.7 dB nHL。Wu等[27]对78例c.109G>A纯合及复合杂合突变儿童进行持续6年的听力随访,发现听力进展速度约为1 dB nHL/年。
综上所述,COL4A5基因及GJB2基因突变所致听力损伤的临床表现具有共性,但目前对该患儿的随访时间短,很难确定该患儿的听力损伤由哪一个基因引起,或者说是共同作用。由此给我们一些启发,确诊某种综合征的患者出现一种临床症状后,我们不单单需要考虑与此疾病相关,还需警惕有无合并其他疾病,以防漏诊误诊。由于GJB2基因也会导致综合征性耳聋,因此我们还需随访监测患儿有无其他症状出现;同时作为AS患者,还需监测其肾功能,动态复查,及时用药,改善患儿预后。
