新生儿糖尿病的诊治思路
2022-05-16王春林卢惠飞
王春林 卢惠飞
浙江大学医学院附属第一医院儿科(浙江杭州 310003)
新生儿糖尿病(neonatal diabetes mellitus,NDM)是指生后6 个月内发病的糖尿病[1]。NDM发病率为1/90000~1/160000[2]。按表型特征可分为暂时性新生儿糖尿病(transient neonatal diabetes mellitus,TNDM)、持续性新生儿糖尿病(persistent neonatal diabetes mellitus,PNDM)和NDM相关综合征。TNDM 患儿症状常在起病后数周或数月内缓解,中位缓解年龄为12周龄[3],缓解期间胰岛β细胞的功能正常[4],但约50%~60%的患儿会在儿童期或青春期复发[1]。TNDM最常见的原因是6q24印记区异常,其次是ATP 敏感性钾通道(KATP)基因变异,包括KCNJ 11和ABCC 8基因。PNDM 患儿需终身治疗,最常见的原因是KCNJ 11(30%~50%)和ABCC8(8%~10%)基因变异,其次是INS基因变异(12%~20%)[5]。目前已知的有30多种单基因NDM相关亚型,不同亚型的NDM临床表现也不同,常以低出生体质量、高血糖、糖尿、多尿、尿频、呼吸急促和脱水为主要特征[6]。
NDM 分型诊断的金标准是基因诊断。临床常用的基因检测有一代Sanger测序法,高通量二代测序(Next-generation sequencing,NGS)包括目标靶向测序(TNGS)、全外显子测序(WES)和全基因组测序(WGS)。目前核心家系全外显子测序(Trio-WES)因其相对阳性率高、经济成本相对低逐渐成为单基因遗传病的首选方案。对于复杂的糖尿病相关综合征,可选用甲基化特异性的多重连接依赖的探针扩增(MS-MLPA)方法或其他检测方法。
1 基因检测技术在NDM诊断中的选择
早期正确诊断和分型有助于实现精准个体化治疗、判断预后,美国糖尿病学会(ADA)建议所有6月龄内发病的糖尿病患儿应立即行基因检测[1],包括自身抗体阳性者[6],因少部分单基因糖尿病患儿自身抗体呈阳性,对于自身抗体阴性的6~12 月龄的婴儿仍建议行基因检测。NDM的基因检测流程见图1[6]。对于有NDM 家族史的孕妇,可以通过羊膜穿刺术或绒毛膜绒毛取样检测胎儿细胞DNA,产前诊断了解胎儿的情况[6]。

图1 NDM 的基因检测流程
基于NDM 临床表型和基因型的异质性以及临床医生对单基因糖尿病认识的差异,我们推荐:①有明确家族单基因糖尿病遗传病史及临床诊断明确的NDM相关综合征(如X连锁内分泌腺病肠病伴免疫失调综合征,IPEX 综合征)进行目的基因一代Sanger测序或者目的基因MLPA技术检测(如6q24甲基化异常的TNDM);②家族中无单基因遗传性糖尿病的NDM或者无明确靶向致病基因的患者,建议进行Trio-WES测序;③对临床高度怀疑单基因变异导致的NDM,而遗传学检测报告“阴性”的患者,需要临床医生与遗传检测实验室相关人员,尤其是数据生物信息分析人员充分沟通,以免“遗漏”。
2 NDM基因型
目前已经报道的NDM 致病基因根据功能可分为3类:①胰腺发育异常:PDX1、PTF1A、HNF1B、PFX 6、GATA 6、GATA 4、GLIS 3、NEUROG 3、NEUROD 1、PAX 6、MNX 1、NKX 2-2;②胰岛β 细胞数量异常:INS、EIF 2 AK 3、IER 3 IP 1、FOXP 3、WFS1;③胰岛β细胞功能异常:KCNJ11、ABCC8、INS、GCK、SLC 2 A 2、SLC 19 A 2[3]。发生糖尿病酮症酸中毒(DKA)风险最高的基因是KCNJ 11和ABCC 8,其他基因如INS、EIF 2 AK 3、FOXP 3、GATA 6、PDX 1等也有报道[7]。NDM 常伴有胰腺外表现,如甲状腺功能减退(GLIS 3)、骨骼畸形(EIF 2 AK 3)、脑畸形(IER 3 IP 1、NEUROD 1、PA X 6、PTF 1 A)、肝功能障碍(EIF 2 A K 3、SLC 2 A 2 A)、尿崩症(WFS 1)、肠闭锁(RFX 6)、腹泻(FOXP 3、NEUROG 3)、胆汁淤积(GLIS 3、RFX6)等[8]。
常见的NDM基因致病机制如下,部分少见基因型及主要临床表现见表1[3,6]。
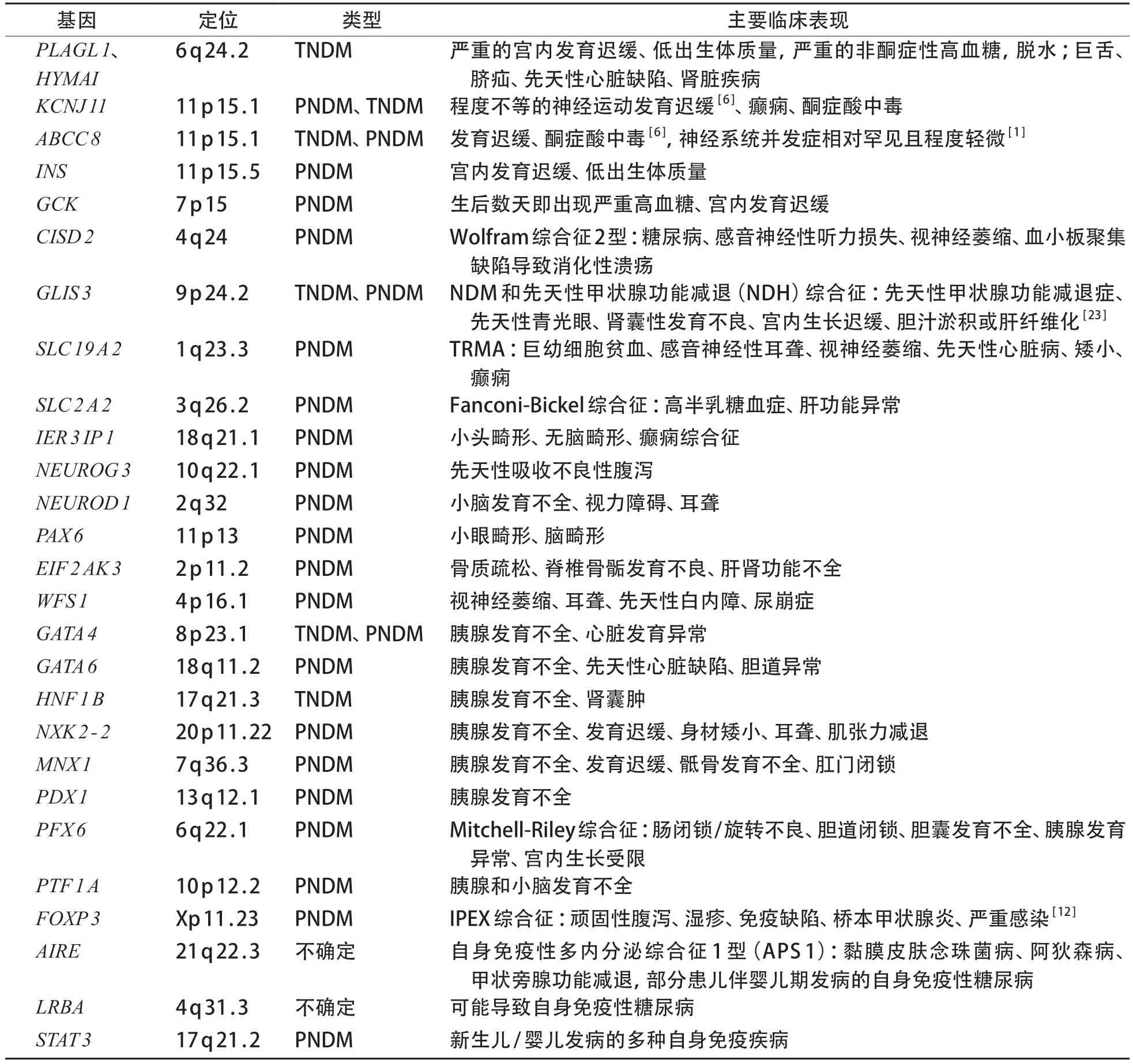
表1 NDM主要基因亚型及临床表现
2.1 6q24印记区域异常
染色体6q24印记区域的基因变异或甲基化异常是TNDM最常见的原因,约占70%[6],以PLAGL1和HYMAI基因变异多见。其分子机制有3种:①6号染色体的父系单亲二倍体;②6q24区域的父系重复;③母亲等位基因的低甲基化[9]。严重的宫内发育迟缓、严重的非酮症性高血糖,巨舌、脐疝、先天性心脏缺陷、肾脏疾病是其典型的临床表型。
2.2 ATP敏感性钾通道(KATP)基因变异
KATP是由Kir6.2亚基和SUR1亚基组成的复合物,它们分别由KCNJ 11和ABCC 8基因编码,基因激活变异导致KATP通道开放、胰岛素分泌减少,导致高血糖,是PNDM 最常见的致病基因,也是TNDM的次要原因。约90%的变异是新生变异,通常没有NDM 家族史,但在家族病例中呈常染色体显性遗传。部分为纯合或复合杂合变异,为隐性遗传,后代多不发病。KCNJ 11变异者中约20%可出现严重神经系统发育异常,表现为明显的发育迟缓、早发癫痫,称为NDM发育迟缓癫痫(DEND)综合征。然而以NDM、轻度发育迟缓、不伴有癫痫为表现的中度DEND综合征更为常见。ABCC8变异者神经系统并发症相对少见且程度轻微。大约90%的KCNJ11或ABCC8变异的NDM患儿口服磺脲类药物治疗血糖控制良好。
2.3 胰岛素(INS)基因变异
INS基因杂合变异是PNDM 的次要原因。以往认为,错误折叠的胰岛素原在内质网中发生降解,导致严重的内质网应激和β细胞凋亡。但最近的研究表明,INS变异并不一定会导致β细胞凋亡,而是慢性内质网应激会干扰β细胞的生长和发育[10]。大多INS杂合变异为散发,约20%有常染色体显性NDM阳性家族史,纯合和复合杂合变异罕见。INS变异患者可在6月龄后起病,多不伴神经系统异常。
2.4 葡萄糖激酶(GCK)基因变异
葡萄糖激酶是胰岛β细胞的葡萄糖感受器,是葡萄糖磷酸化过程的关键酶。GCK基因杂合变异导致青少年起病的成人型糖尿病2型(maturity-onset diabetes of the young type 2,MODY2)。但在纯合子或复合杂合子中,由于完全缺乏葡萄糖激酶,导致NDM。GCK基因变异占PNDM的2%~3%,呈隐性遗传。若先证者父母表现为无症状轻度血糖升高,应考虑该疾病,并建议父母监测空腹血糖。临床表型为严重高血糖及宫内发育迟缓,通常生后数天即出现高血糖表现。除糖尿病外,无其他胰腺外特征。
2.5 GATA6基因变异
GATA6基因的杂合子变异是合并胰腺发育不全的NDM 中最常见的原因。其致病性变异与多系统先天性缺陷相关,特别是心脏发育异常和胰腺发育不全。其他包括胰腺外分泌功能不全、胆道闭锁、甲状腺功能减退、垂体发育不全/垂体功能低下、肾脏结构异常、神经认知缺陷和癫痫等[11]。
2.6 相关综合征
Wolcott-Rallison综合征是由EIF2AK3基因纯合或复合杂合变异导致的一种罕见的综合征,呈常染色体隐性遗传。该基因编码一种参与调节内质网应激反应的蛋白,错误折叠的蛋白质在内质网积累,最终诱导胰岛β 细胞凋亡。同胞发病风险为25.0%,在父母非近亲结婚的情况下,后代发病风险非常低。临床以早发性糖尿病、脊椎骨骺发育不良、复发性肝肾功能不全为特点。婴儿期糖尿病多为首发表现,其他临床表现可能到3~4岁才出现,PNDM需考虑该综合征,尤其是父母为近亲结婚者,尽早行基因检测。
IPEX 综合征(X 连锁内分泌腺病肠病伴免疫失调综合征)由FOXP3基因变异所致,是唯一确定的与β细胞自身免疫和胰岛自身抗体相关的PNDM。临床表现为内分泌疾病(PNDM、桥本甲状腺炎等),免疫缺陷,皮肤损害,肠病以及危及生命的感染[12]。
Mitchell-Riley综合征是由染色体6q22上RFX6基因纯合或复合杂合变异引起的。该基因在肠、胆囊和胰腺β细胞的发育中具有重要的作用。临床表现为新生儿糖尿病、肠闭锁/旋转不良、胆道闭锁、胆囊发育不全、胰腺缺失或异常和宫内生长受限等。糖尿病大多在生后2 周内被诊断,少数患儿在儿童期出现糖尿病。预后较差。
3 诊断
新生儿无法表达任何症状,临床表现隐匿、不易察觉,故新生儿糖尿病的诊断有时是困难的。新生儿期或生后6周内出现持续空腹血糖≥7 mmol/L(120 mg/dL),病程持续2周以上,需予胰岛素治疗维持正常血糖者要诊断NDM。NDM 诊断流程图见图2。对疑似患儿的初步评估包括血糖、C 肽、胰岛素、尿酮和胰腺超声等检查。由于受到母亲抗体的影响,糖尿病自身抗体检测对6月龄内诊断NDM的患儿不是必要的,6月龄后的患儿建议尽早完善自身抗体检测,有助于糖尿病的分型,如IPEX 综合征可出现自身抗体阳性。NDM 需排除应激,感染,糖皮质激素或儿茶酚胺的使用,过量的葡萄糖输注等其他原因引起的高血糖,尤其需与短暂性特发性高血糖(transient idiopathic hyperglycemia,TIH)鉴别。TIH主要发生在妊娠30周前出生的婴儿生后第一周或出生体质量≤1 000g,主要接受肠外营养者[13]。与TIH 相比,NDM 的特征是血糖值更高,需更早予胰岛素治疗,胰岛素需求量更高,治疗时间更长[13]。NDM常以DKA为首发表现,注意与感染性休克、遗传代谢病危象等鉴别。
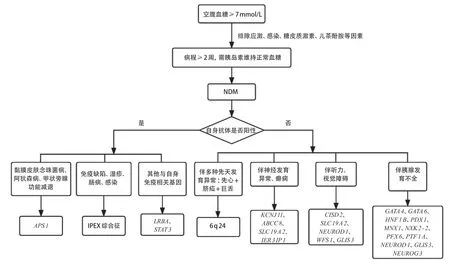
图2 NDM 诊断流程图
4 治疗和管理
NDM 的治疗和管理是儿科医生面临的一大挑战。由于新生儿大脑的脆弱性,高血糖或低血糖对神经发育和认知表现均有损害,因此维持适当的血糖水平和选择最佳的治疗对于NDM 患儿的正常生长和神经发育至关重要。
4.1 胰岛素替代治疗
大多数新诊断为NDM 的患儿早期需予胰岛素治疗[14]。目前暂无NDM的治疗指南,有文献表明,当血糖>180 mg/dL时,应进行干预,然而由于胰岛素干预后低血糖的风险高于短期高血糖的风险,故相关研究建议血糖水平>500 mg/dL时予胰岛素治疗[2]。但是,当血糖水平>360 mg/dL时,由于渗透压的变化,可能导致颅内出血[2]。因此我们建议当血糖水平持续超过250 mg/dL时予胰岛素干预[2]。胰岛素的治疗原则以及方案可参考儿童及青少年糖尿病的胰岛素治疗指南(2010年版)[15]。
4.2 磺脲类药物口服治疗
大约90%的KCNJ11或ABCC8变异的NDM患儿口服磺脲类药物治疗,可达到良好的初始血糖控制。年龄越小、病程越短的患儿予磺脲类药物治疗还可逆转部分KCNJ 11变异导致的神经发育迟缓,同时可增加从胰岛素转换至磺脲类药物治疗的成功率[16]。一项为期10年的多中心、国际队列随访研究显示磺脲类药物治疗KCNJ 11变异的PNDM 患儿是非常有效和安全的[17]。部分患儿加用磺脲类药物后可减少胰岛素剂量,但不能完全停用,建议继续予磺脲类和胰岛素联合治疗[16]。如果在加用磺脲类药物后C 肽水平、胰岛素剂量没有变化,建议继续予磺脲类药物治疗3 个月,若未发现对中枢神经系统受益,则可以停用磺脲类药物[16]。以格列本脲为例,平均有效治疗剂量为0.5 mg/(kg·d),最大可达2.3 mg/(kg·d)[1]。磺脲类药物最常见的副作用是腹泻,恶心和低血糖在年龄小的患儿中也有报道,但这些副作用是暂时的,通常不需要中断治疗。
4.3 其他治疗
硫胺素反应性巨幼细胞贫血(thiamine-responsive megaloblastic anemia,TRMA)综合征除予外源性胰岛素治疗高血糖外,有研究报道,补充硫胺素可使大多数患儿停用胰岛素或减少剂量[18],并可能延迟糖尿病的发病[19],然而由于β 细胞凋亡加速,一些患者可能在青春期需予胰岛素治疗[18]。IPEX 综合征急性自身免疫期可予免疫抑制剂治疗,异基因造血干细胞移植是IPEX 综合征的首选治疗,是唯一可行的长期生存的选择[20]。病例报道部分IPEX 综合征患儿移植后糖尿病情况并无改善,但也有患儿糖尿病情况有所改善,提示部分患儿可能存在胰岛细胞可逆或不完全损伤[21]。基于干细胞的治疗被认为是一种有前途的糖尿病治疗方法,但目前仍有许多问题和技术障碍有待解决[22]。
5 结语
NDM 的临床表现不典型,起病隐匿,容易延误诊断,另一方面,NDM的诊断相对容易,诊断标准清晰明确,但NDM 的分型、遗传学诊断却较困难。临床医生提供尽可能全面的临床表现,选择合理的遗传学检测技术,可早期明确NDM的遗传学分型,有助于达到精准治疗。NDM 的治疗和管理比较棘手,除KCNJ11或ABCC8变异的NDM患儿可以口服磺脲类药物治疗达到良好的血糖控制外,绝大部分的NDM 患者需要胰岛素替代治疗,遵循饮食管理、血糖监测、胰岛素替代治疗、合理的运动以及健康教育等“五驾马车”综合管理原则,但在新生儿阶段实施困难更大,更具有挑战性。
