一个遗传性非综合征型耳聋大家系的GSDME基因突变分析
2022-05-16黄演林余丽华丁红珂曾玉坤刘玲陈洽鑫刘畅
黄演林 余丽华 丁红珂 曾玉坤 刘玲 陈洽鑫 刘畅*
(1.广东省妇幼保健院 医学遗传中心,广东 广州 511400;2.广东省妇幼保健院 耳鼻喉科,广东 广州 511400)
耳聋是由听觉系统中的听神经及各级中枢病变而引起的听觉障碍,是最常见的人类感觉系统缺陷[1]。先天性因素(遗传)和后天性因素均可导致耳聋。据调查,世界范围内约4.66亿人患中度以上听力残疾,我国听力言语残疾者达2780万,约占中国残疾人总数的33%,居我国各类残疾之首[2]。非综合型耳聋是指除听力障碍外不涉及外耳畸形或其他器官系统的异常表现,约80%为常染色体隐性遗传,20%为常染色体显性遗传[3]。目前,已发现超过25个基因与常染色体显性非综合征型听力障碍相关,大部分为晚发型听力障碍[4]。常染色体隐性非综合征型障碍具有较大的遗传异质性,在大多数人群中,重度至极重度常隐非综合征型听力障碍的最常见原因是GJB2突变,轻度至中度常隐听力障碍的最常见原因是STRC突变[5]。
由于非综合征型耳聋存在临床表现的差异及致病基因的不明确性,Sanger测序不适用于非综合征型耳聋的基因诊断。在二代测序技术(next-generation sequencing,NGS)的推动下,耳聋基因NGS检测包发展迅速,可通过耳聋相关基因的目标序列捕获,进行高通量测序,达到很高的灵敏度和准确度,为耳聋分子诊断提供了新的方法[6]。本研究利用遗传性耳聋精选基因检测包对一耳聋先证者进行基因检测,并对检测到的突变位点进行Sanger测序验证,同时收集家系其他成员进行Sanger测序。不仅成功鉴定了一种导致Joubert综合征的新突变,丰富了Joubert综合征的病因信息,同时为罕见疾病的产前诊断及临床确诊提供了一套可行的新方法。不仅成功鉴定了一种导致晚发型非综合征型耳聋的致病变异,丰富了遗传性耳聋的病因信息,同时为耳聋分子诊断提供一套可行的新方法。
1 材料与方法
1.1 病历资料 谭CY,XX岁,因“家族中有多人耳聋”于广东省妇幼保健院产前诊断中心就诊。家族史:本人及父母、哥哥、大姑、二姑均体健,未见听力障碍,其爷爷、三姑和小姑患有耳聋多年;曾爷爷也患有耳聋(已故),其姑婆、大表姑、二表叔和小表叔均患有耳聋,二表叔女儿为耳聋成员中年龄最小者(21岁),患有轻度听力损失,详见该家系图(图1)。本研究共收集该家系36个成员(3人已故)的资料,由于就诊者本人及父母均无听力障碍表现,首次实验室检查抽取其三姑谭JZ(44岁,耳聋30年)外周血,行遗传性耳聋精选基因包检测,测序结果经Sanger测序验证。耳聋基因包检测及Sanger验证结果均显示存在致病基因位点,后收集该家系33人外周血进行Sanger测序。本研究获得了广东省妇幼保健院机构审查委员会及广州医科大学伦理委员会的批准。所有样品的采集均取得了参与者的书面知情同意书。
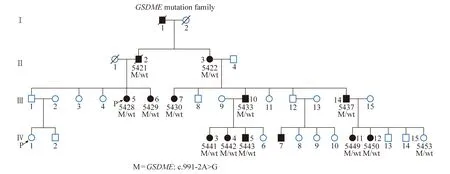
图1 GSDME基因突变致聋大家系图
1.2 实验方法
1.2.1 外周血DNA提取 用QIAGEN公司基因组DNA提取试剂盒QIAGEN DNA Blood Mini Kit按试剂盒说明书提取外周血DNA,使用Nanodrop2000超微量分光光度计进行提取DNA的定量,要求DNA样本纯度OD A260/280介于1.8~2.0之间。
1.2.2 遗传性耳聋基因包检测与分析 质检合格的基因组DNA经超声破碎仪打断、末端修复、扩增、纯化等操作制备测序文库,采用特异性的捕获探针(Roche NimbleGen, Madison, WI, USA)杂交富集目标区域的DNA序列,目标区域包括204个耳聋相关基因,3148个编码区总共含有497507个碱基。采用Illumina NovaSeq 6000平台进行测序。应用商业软件SoftGenetics NextGENe V2.4.1.2进行数据分析和变异检测。参考基因组版本为GRCh37/hg19。根据gnomAD数据库中次等位基因频率(MAF)进行变异初筛(即过滤掉MAF>0.01的变异,GJB2 NM004004.5:c109G>A等高频致病位点除外)。采用VarCards在线预测网站(包含SIFT、Polyphen2、MutationTaster和REVEL等23种预测算法)以及SpliceAI、varSEAK等工具对变异进行有害性预测,结合ClinVar、HGMD、Varsome和变异类型、文献病例报道等信息,并根据ACMG/AMP指南对变异进行综合评级和分类[7]。本文GSDME基因使用的转录本为NM_001127453.2。
1.2.3 Sanger测序 根据高通量测序结果用Sanger测序对先证者DNA及其他33个家系成员外周血DNA进行突变位点的检测和验证。采用Primer5软件设计引物,扩增GSDME基因第8号外显子及上下游10个bp的序列。引物序列为:GSDME-E8F:CACCAAGGATTAGCAATTTCAG,GSDME-E8R:AGAAGGGAAGGACCTGTAT。PCR扩增条件为:95℃预变性5min;95℃变性30s,58℃退火30s,72℃延伸30s,共40个循环;延伸72℃ 5min。
2 结果
2.1 二代测序结果 先证者耳聋基因包检测及Sanger验证结果显示:GSDME基因第7号内含子上(距离第8号外显子2bp)发生了碱基替换,即c.991-2A>G,形成剪接位点变异,为杂合突变,通过外周血Sanger测序证实,该变异来源于先证者父亲,其父亲也为耳聋患者(先证者母亲已故,未行基因检测,生前未见耳聋)。变异信息见表1。GSDME基因相关疾病为常显遗传性耳聋5型,是常染色体显性遗传。临床表现主要为语后、高频率、进行性听力损伤,一般在第一个10年发病[8]。

表1 GSDME突变家系二代测序结果
2.2 Sanger测序结果 本家系33个成员针对GSDME:c.991-2A>G致病位点进行了Sanger测序。结果显示:所有耳聋家系成员均检出该杂合致病位点(8/33,24.2%),所有听力正常者均未检出(25/33,75.8%),即约1/4的家系成员患病,Sanger测序结果详见图2。其中,首次就诊者的表妹虽检出该剪接位点变异,但由于其年龄相对较小,目前仅有轻度的听力损失,符合该病进行性听力损伤的特点。
3 讨论
遗传性非综合型耳聋是指除听力障碍外不涉及外耳畸形或其他器官系统的异常表现,约80%为常染色体隐性遗传,20%为常染色体显性遗传[8],目前, 已发现超过25个基因与常染色体显性非综合征型听力障碍相关,大多数会导致语后听力障碍,如ACTG1、CCDC50、EYA4、COL11A2、DIAPH1、MYH9、GSDME、MYO1A、MYO6等等,一般在生后第一个10年内发病,以高频率、进行性听力损失为特征[4]。GSDME基因相关耳聋为常显遗传性耳聋5型(deafness, autosomal dominant 5,DFNA5; OMIM#600994)是一种常染色体显性遗传疾病,是由染色体7p15.3上的GSDME基因发生杂合突变引起的。GSDME基因包含10个外显子,跨度约为60kb[9]。GSDME编码Gasdermin-E蛋白,该蛋白是将非炎症性细胞凋亡转化为细胞焦亡的成孔蛋白的前体,在进行蛋白切割时,释放的N末端部分(Gasdermin-E,N端)与膜结合并形成孔,引发细胞焦亡[10]。DFNA5患者多发病于儿童早期。此疾病是进行性的,临床症状表现为感知性耳聋,双耳在高频率声波波段听力受损,随着时间推移,听力损失扩大到较低频率声波波段并迅速加重[8]。

图2 GSDME基因Sanger测序峰图(传代数和成员编号与家系图一致)
本文我们描述了一个包含36个成员的超大家系,首先对“耳聋30年”的先证者行遗传性耳聋精选基因包检测,发现一个剪接位点致病变异GSDME:c.991-2A>G,后针对33个成员进行了Sanger测序验证,共发现8个家系成员携带该致病位点,且8位成员有7为重度或极重度耳聋,年龄最小的携带者则表现出轻度听力损失,符合GSDME基因异常引起的DFNA5特征。已故的3个成员中有一个亦是耳聋患者。另有25位家系成员未检出该剪接致病变异,且均未耳聋表现。所有阳性位点均为杂合突变,符合孟德尔显性遗传的特点。该家系为GSDME:c.991-2A>G位点所报道的最大家系,同时也是GSDME基因突变所报道的最大家系,家系中包含的患者人数最多(9个)。
本研究发现的位点GSDME:c.991-2A>G是一个新型的剪接位点变异,位于mRNA剪接区域,核苷酸序列高度保守,并且多种计算辅助算法也预测这个变化可能会影响剪接功能。这个变异在中国一个晚发型进行性非综合征耳聋家系的多个患者中被检出,临床表现为高频听力受影响,并随着年龄增长而加重,突变携带者有表现差异性[11]。到目前为止,这个变异在我们的参考人群基因数据库(gnomAD)中未见人群频率记录。综上所述,进一步结合送检者的临床表现和家系分析,依据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)变异分类指南,这个变异暂定为“Ⅱ类-可能致病”。即具有较为充分的证据表明该变异是引起该家系成员耳聋的病因。
综上所述,本研究利用二代测序技术成功鉴定了一种导致晚发型非综合征型耳聋的剪接位点变异,并利用Sanger测序技术,从33个家系成员中确诊了8个DFNA5患者,并且根据变异致病性分析,确认此突变即为上文所述家系的遗传学病因。此家系为迄今报道的最大家系,丰富了遗传性耳聋的病因信息。二代测序技术为耳聋分子诊断提供一套可行的新方法。
