大豆荚皮厚性状QTL定位及候选基因挖掘
2022-05-13王乔蒋洪蔚谢建国潘文婧郑海洋侯立龙熊心武小霞
王乔,蒋洪蔚,谢建国,潘文婧,郑海洋,侯立龙,熊心,武小霞*
(1.东北农业大学,黑龙 江哈尔滨, 150030;2.吉林省农业科学院,吉林省长春市邮编 130033)
大豆荚皮,是影响大豆产量的重要性状。作为大豆生产的主要副产品,大豆荚皮和大豆籽粒的比例达高达1∶2[1]。大豆荚皮中富含大量的蛋白质和纤维素,具有较高的饲用价值[2]。Sruamsiri 等利用大豆荚皮替代部分稻草饲喂奶牛,结果表明奶牛的采食量和4%标准乳产量均有升高[3]。但大豆荚皮的利用率较低,大多数作为农业废弃物进行焚烧,对环境造成极大破坏[4~6],也不利于资源的可持续发展。大豆荚皮厚与大豆单株产量呈负向相关。营养物质在荚部的分配主要集中于荚皮与子粒,荚皮过厚不利于子粒营养物质的积累,降低了可食用率[7]。故大豆荚皮厚的研究对大豆增产、资源再利用等都具有重要意义。
现如今,随着测序技术的发展和新型标记的开发,有关QTL 的研究已经趋于成熟。其中有关QTL的算法,有复合区间作图法(CIM)和完备区间作图法(ICIM),这两种方法都可以精确定位QTL。李永亮等[8]利用CIM 和ICIM 两种方法结合,对来自CSSLs 群体的62 个单株和RIL 群体的147 个单株进行大豆冠层性状QTL定位分析,最终检测到31个相关的QTL 位点。姚丹等[9]以ICIM 法对吉农18 和吉育47 杂交后代F2及次级分离群体进行大豆油分相关QTL 定位,共检测到分布于4 条染色体的7 个与高油含量相关的QTL 位点。ICIM 法简化了CIM 法中由背景遗传调控的变异,精细了对QTL 的检测效率[10]。大豆荚皮厚受环境因素和遗传背景的共同调控,因此也是由多基因控制的数量性状。我们利用完备区间作图法(ICIM)对其进行QTL定位。
集群分离分析法(BSA)利用遗传群体构建高低混池进行高通量测序,找到与目标性状相关标记,从而实现QTL 精细定位。这是一种对遗传群体的后代筛选极端表型,以构建DNA 混池,快速检测与目的性状相关联分子标记的方法[11]。BSA 最早由Michelmore[12]于1991 年提出,主要用于鉴定基因组某个区域内的标记。现在,BSA 混池测序通过开发分子标记,广泛应用于遗传学、农艺基因组学、分子标记辅助育种[13]。随着全基因组高通量测序技术的发展,利用BSA 分析与全基因组测序相结合的BSA混池测序技术,被广泛应用到QTL 定位和候选基因挖掘[14]。人们通过使用较大的遗传群体,增加极端表型个体数量和提高分子标记密度,使BSA 更适用于靶基因定位,从而无需通过使用阳性标记对整个群体进行基因分型验证假定标记[15]。Shen 等[16]利用BSA混池测序和基于SSR标记所得的QTL定位出三个小麦镰刀菌枯萎病(FHB)抗性区间。Trick 等[17]将测序数据与BSA 分析相结合,对小麦的谷物蛋白含量进行精细定位,成功将基因GPC-B1定位在0.4 cM 的区间内。Song 等[18]用基于下一代测序(NGS)的BSA 法,快速定位植物中两个控制大豆种子子叶颜色的基因。
本实验首先构建由SN14 与ZYD00006 为父母本的CSSLs 群体,然后利用ICIM 进行QTL 定位,共获得位于6 条连锁群的13 个QTL 区间。同时构建高低混池,进行BSA 混池测序,得到新的QTL 区间。对两方法得到的QTL 区间进行整合,得到共识QTL区间。对该区间内的所有基因进行注释,通过基因的序列对比、氨基酸变异确定候选基因。最终得到与大豆荚皮厚相关的目的基因,为高产大豆品种的遗传改良奠定理论基础。
1 材料与方法
1.1 材料
从2006 年至2018 年,使用母本黑龙江省栽培品种绥农14(SN14)为与父本野生品种ZYD00006进行杂交,再经过多次的回交、自交,最后构建一套包含208 个株系的染色体片段替代系(CSSLs)[19]。每个品系每年种成株行,行距60 cm,株距6 cm,行长5 m。材料成熟后,每行材料随机选取5 株,每株材料随机取十个荚皮,整齐叠放在一起,用游标卡尺进行测量,获取荚皮厚的表型数据[20]。
1.2 BSA(Bulked Segregant Analysis)混池的构建
根据2013至2016荚皮厚4年的表型数据,计算平均值,整体表型呈现标准的正态分布(图1),极大值2.40 cm 和极小值1.54 cm,相差0.86 cm,变异程度较大,以平均值加减一倍标准差筛选出极端表型各30 份材料(如表1),构建DNA 混池,为混池测序做准备。

表1 荚皮厚高低混池的材料组成Table 1 Phenotypic screening of pod thickness in high and low mixed ponds
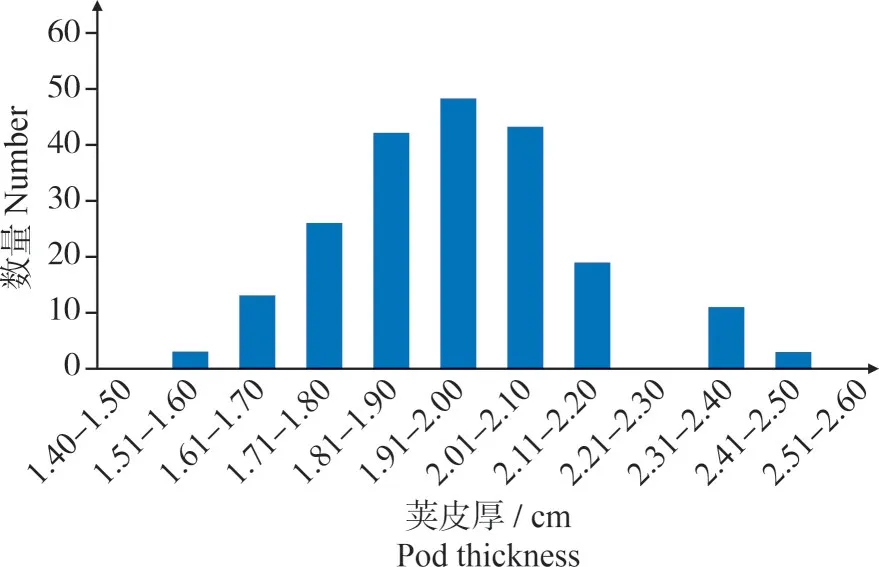
图1 CSSLs群体荚皮厚四年平均值表型数据Fig.1 Four-year average data of pod thickness of CSSLs population
1.3 混池测序数据处理
选取新鲜的叶片,使用CTAB 法提取植物组织的DNA,对提取的DNA 片段进行修饰,添加测序接头。利用PCR扩增对DNA 片段进行富集,完成测序混池的构建。最后合格的DNA 文库,送于Illumina-HiSeqTM测序平台进行测序。为了保证数据分析的可靠性,对原始的测序数据进行以下处理:去掉测序接头,过滤数据中未检测碱基数量比值大于10%的数据,再次过滤数据中低质量碱基——即碱基质量值小于10%大于50%的数据,使用GATK 软件进行局部重新比对,过滤得到高质量的SNP 位点。最后利用BW[21]软件将测序数据定位到参考基因组中,进行变异分析。
1.4 统计与分析
对CSSLs群体荚皮厚性状的四年表型数据进行统计分析。利用ICIMapping4.1 对CSSLs 群体中“BIP”模型进行QTL 分析。以LOD>2.5 为标准,确定QTL 存在的依据。使用MapChart2.2 软件,将定位得出的QTL 标注在连锁群上。QTL 命名方法如下:q+性状名称+LG或LG编号+“-”+QTL编号[8]。
1.5 SNP-index关联分析
SNP-index 是通过计算极端混池间基因型频率的显著差异,进而进行关联分析[22~24],利用双亲的重测序结果,计算极端混池SNP-index,寻找与性状紧密相关的位点。计算公式如下:
SNP-index(Mut)=ρx/(ρX+ρx)
SNP-index(WT)=ρx/(ρX+ρx)
ΔSNP-index=SNP-index(Mut)-SNP-index(WT)
1.6 基因注释
以Williams 82.a2.v1 为参考基因组,根据所得的共识QTL 区间,利用NCBI 数据库对目标区间的所有基因进行注释。结合生物信息学分析,进行序列比对,同源基因功能查找,分析氨基酸变异,预测候选基因,确定与荚皮厚相关的目的基因。
2 结果与分析
2.1 CSSLs群体荚皮厚表型数据分析
对CSSLs 群体2013 年至2016 年四年的荚皮厚表型数据(表2)统计分析。四年间的表型数据的极高值3.62 cm、极低值1.23 cm,相差2.39 cm,变异较大。四年间表型的平均值差异也较大。2013 年群体荚皮厚的平均值为1.67 cm,与2016 年群体表型的平均值1.67 cm 较接近,而同比明显低于2014和2015荚皮厚表型平均值。CSSLs群体荚皮厚性状在4个环境的数据分布都显示为典型的近似正态分布,说明荚皮厚性状符合数量性状遗传特征,适用于QTL 定位分析(图2)。
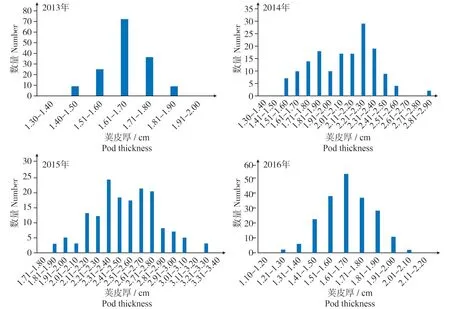
图2 CSSLs群体四年荚皮厚表型数据频率分布Fig.2 Frequency distribution of pod thickness phenotype data of CSSLs population in four years

表2 四年CSSLs群体的荚皮厚表型数据参数Table 2 Phenotypic parameters of pod thickness in four year CSSLs population
2.2 荚皮厚性状QTL分析
我们利用ICIM 法对大豆荚皮厚进行QTL 定位,在8个连锁群上共定位到13个与荚皮厚相关的QTL(表3,图3)。表型贡献率为2.98%~14.44%,LOD 值在2.53~8.43 之间。F 连锁群上被检测到的QTL 最多有3 个,分别为qPT-f-1、qPT-f-2和qPTf-3。三个QTL位点的区间位置非常接近,可能是紧密连锁的关系。N、K 连锁群上的QTL 为2 个,其他的连锁群仅有1 个。其中qPT-n-2在2015 年和Mean两个环境中同时被检测到,重复被检测到也说明了该QTL位点的可靠性。

图3 CSSLs群体荚皮厚性状QTL分析Fig.3 QTL analysis of pod thickness in CSSLs population

表3 ICIM法对大豆夹皮厚性状QTL分析Table 3 Analysis of QTL for pod thickness in soybean by ICIM
2.3 BSA(bulked segregant analysis)测序数据分析
进行DNA 混池测序的极端材料中,共检测到1 953 662 个原始SNP。经过SNP 过滤,最终得到1 630 948个高质量的SNP位点。
利用混池间的基因型频率和荚皮厚进行关联分析,以群体的中值+3 倍标准差为阈值,计算每个位点的ED 值,选择ED 值大于阈值的区域为候选区间。最终在4 个连锁群上定位了7 个与大豆荚皮厚相关的候选区域(表4,图4)。其中,F连锁群上检测到3个QTL位点,N连锁群检测到2个QTL位点。这与ICIM 法在F连锁群和N连锁群检测到3个QTL和2个QTL的结果一致,说明F连锁群和N连锁群含有调控大豆荚皮厚性状的QTL 位点。最小的QTL 区间qPT-f-4仅为0.08 Mb,最大的QTL 区间qPT-b1-1达到8 Mb。
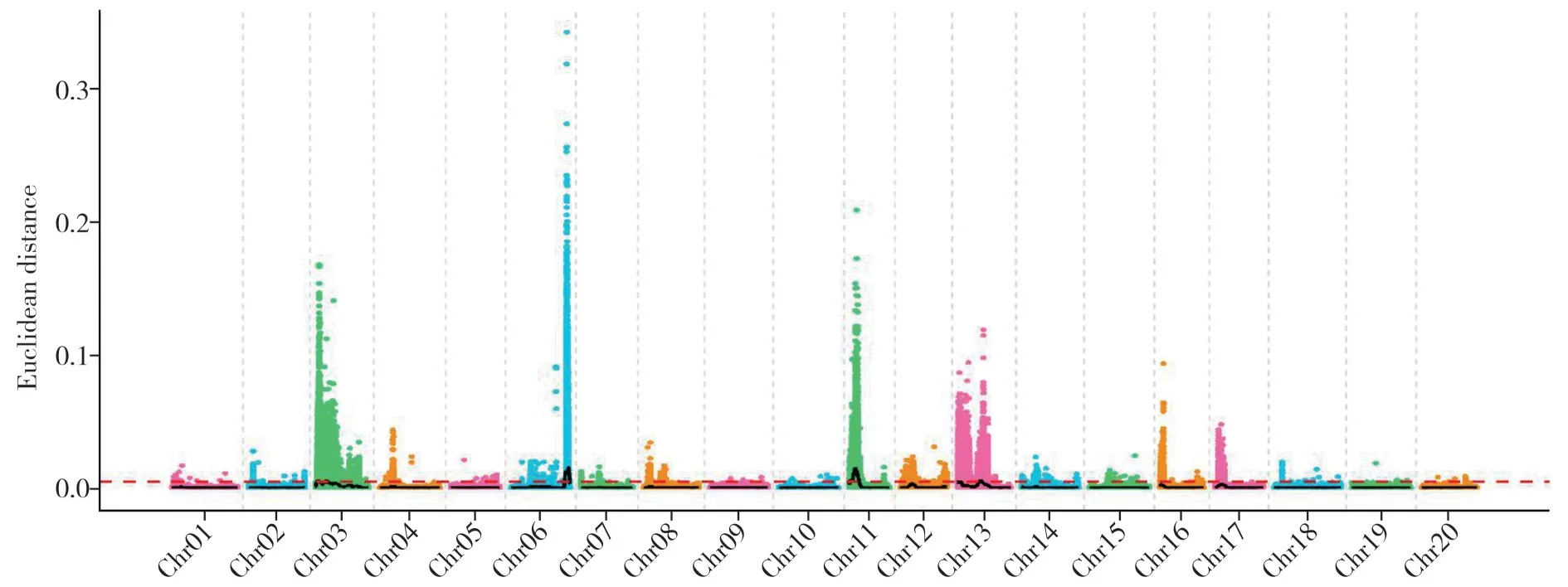
图4 CSSLs群体荚皮厚性状BSA分析Fig.4 BSA analysis of pod thickness in CSSLs population
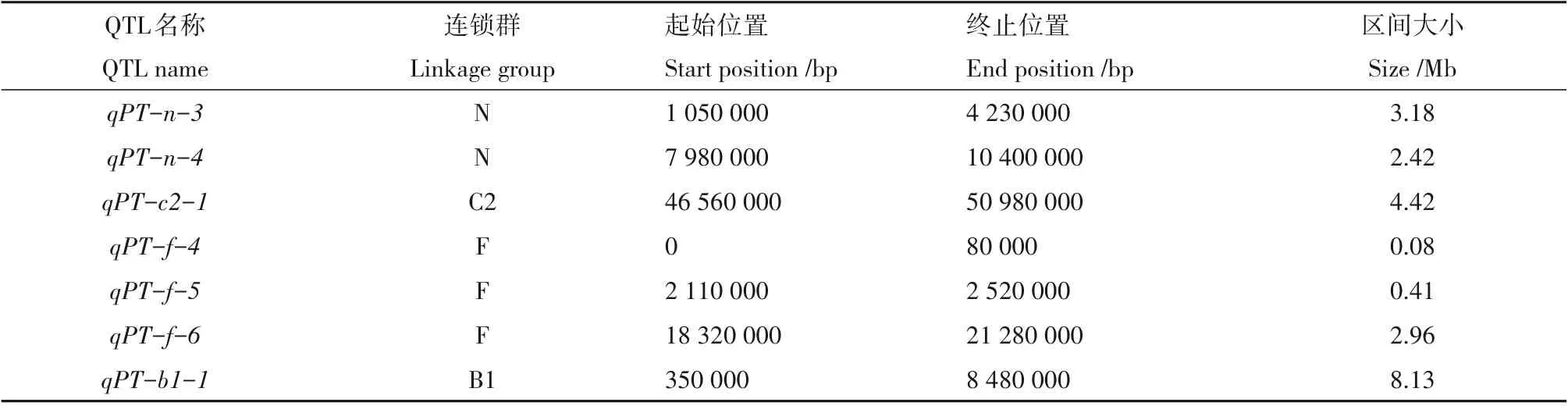
表4 BSA法对大豆荚皮厚性状QTL分析Table 4 Analysis of QTLs associated with pod thickness in soybean by BSA
2.4 共识QTL区间
我们通过比较BSA 法和ICIM 法定位的QTL 位点,找到公共的QTL 区间。这样既能得到相对稳定的QTL 位点,也能缩小了QTL 的置信区间。将ICIM法检测到的13 个QTL 区间与BSA 法检测到的7 个QTL 位点相结合(图5),获得1 个稳定的QTL 位点qPT-n-2。同时,qPT-n-2在多个环境中同时被检测到,说明该QTL 是调控大豆荚皮厚的稳定可靠QTL 位点。最终通过两种分析方法,共同将影响大豆荚皮厚性状的候选区间精确到3 号染色体0.03Mb(2 970 674 bp~3 000 004 bp)的区间内,为后续候选基因的挖掘工作奠定基础。
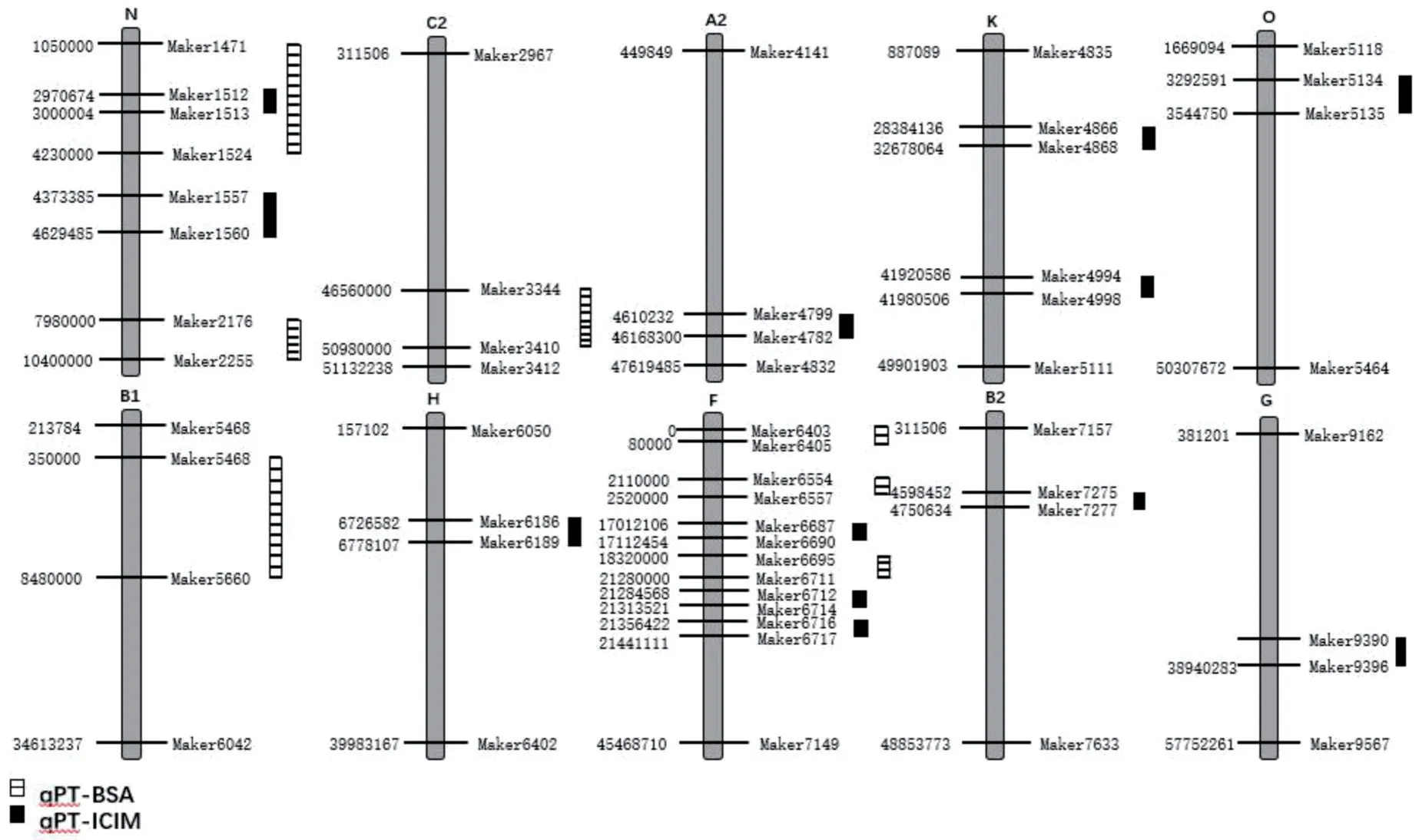
图5 CSSLs群体荚皮厚性状QTL在连锁群上的分布Fig.5 Distribution of QTLs about pod thickness of CSSLs populations on linkage groups
2.5 候选基因分析
为得到共识QTL 区间内的功能基因,我们对该区间进行了基因注释,得到4 个候选基因Gly⁃ma. 03G027000、Glyma. 03G027100、Glyma. 03G0272 00、Glyma. 03G027300注 释 结 果(表5)。Gly⁃ma.03G027000的拟南芥同源基因为AT3G50120,是未知功能的植物蛋白;Glyma. 03G027100没有拟南芥同源基因,其GO 注释为膜的组成部分;Gly⁃ma.03G027200的拟南芥同源基因为AT1G35710,注释为具有富含亮氨酸重复域的蛋白激酶家族蛋白,其GO 注释为蛋白质磷酸化以及蛋白激酶活性等功能。Glyma. 03G027300的拟南芥同源基因为AT4G00110,注释为UDP-D-葡萄糖醛酸4-表异构酶3。通过Soybase 公共数据库查询,四个基因在花、荚不同时期的表达量差异较大。Glyma.03G027 000和Glyma.03G027200在花荚各个时期表达量都非常低,而Glyma. 03G027100和Glyma. 03G027300在花荚各个时期的表达量则显著的较高,尤其是Glyma. 03G027300在花和荚中的表达量最高(如图6)。

图6 soybase数据库候选基因花荚时期表达量分析Fig.6 Expression analysis of candidate genes at flowering and pod stage in soybase database
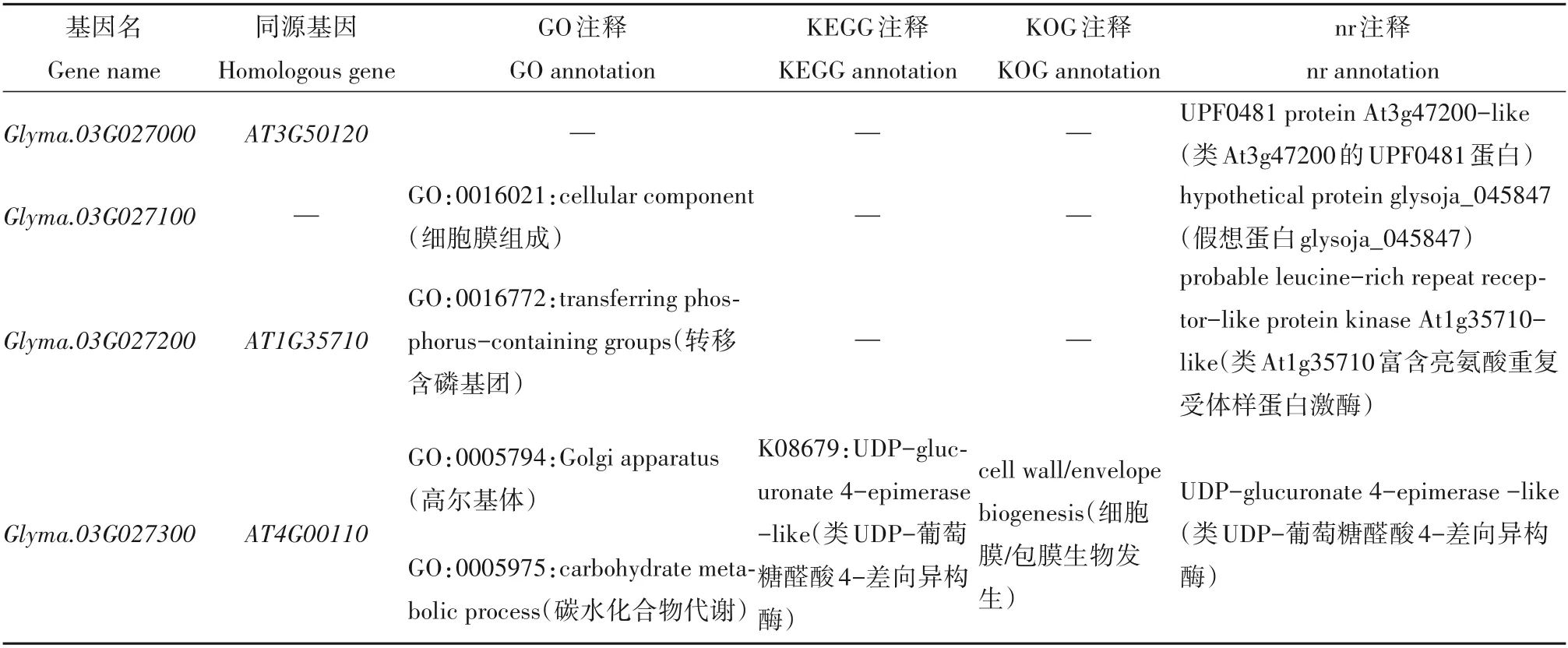
表5 大豆荚皮厚性状候选基因注释信息Table5 Candidate gene annotation information related to pod thickness of soybean
2.6 候选基因氨基酸序列比对
通过比对SN14和ZYD00006两个亲本间的碱基和氨基酸序列,Glyma. 03G027200的碱基变异程度很低,只有两个碱基发生突变,对应的组氨酸和亮氨酸氨基酸发生了非同义突变,氨基酸的相似度为99.82%。Glyma. 03G027100和Glyma. 03G027300的基因碱基序列变异程度很高。Glyma.03G027100有26处碱基发生突变,氨基酸序列有17种非同义突变,Glyma. 03G027300的碱基突变为9 处,对应9 处氨基酸都发生了非同义突变。
3 讨论与结论
现阶段对大豆荚皮厚性状研究内容较少。但是大豆荚皮厚与大豆的产量和抗病虫能力都紧密相关,是未来大豆专化目标育种及分子育种的重要性状。
如今对于大豆QTL 定位的研究非常多。截止到2020 年1 月,Soybase 数据库(https://www.soybase.org)公布的与大豆蛋白质含量相关的QTL 超过200个,但具有重演性的QTL 很少[25,26]。究其原因是由于数量性状遗传机制复杂,受环境影响较大。传统的QTL 定位需要构建遗传群体,与整个群体全部分析相比,BSA方法与成熟的测序技术相结合,为快速基因定位和基因挖掘提供了技术支持,并为鉴定和开发新的分子标记开辟了一条捷径[27]。由于该方法仅仅分析子代群体中具有极端表型的一些个体,极大地降低了测序成本,而其统计能力也与QTL 定位相当,具有省时省力,精确度高的优点[28,29]。本研究通过ICIM 法和BSA混池测序相结合的方法,共检测到20 个QTL 位点,分别位于N、C2、A2、K、O 等10 条连锁群。位于F 连锁群的QTL 位点有6 个,位置紧密相连,表明F 染色体含有调控大豆荚皮厚性状的重要基因。qPT-n-1位于两种方法检测的共识QTL区间,同时在两个环境中同时多次被检测到,具有重演性、稳定性的特点,故而是和大豆荚皮厚关联的关键QTL。
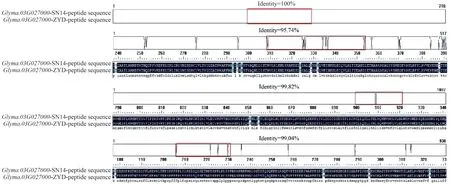
图7 候选基因亲本序列氨基酸比对Fig.7 Amino acid alignment of candidate gene between two parents
本研究对定位到的QTL 区间qPT-n-1内基因进行了分析。结合基因注释信息分析与Soybase 数据库网站提供的基因空间表达分析,最终从中筛选出2 个与大豆荚皮厚性状相关的目的基因,为Glyma.03G027100和Glyma.03G027300。Glyma. 03G027100的GO 注释为细胞膜的组成部分相关,很有可能是调控细胞膜生长的重要功能基因,在荚中高表达,说明与荚皮厚性状密切相关,可能是直接调控大豆荚皮厚性状的基因。Gly⁃ma.03G027300是花和荚中的各个时期表达量最高的基因,该基因的拟南芥同源基因为AT4G00110,在UDP-D-葡萄糖醛酸-表型异构酶的编码中起作用,UDP-葡萄糖是由蔗糖降解所得到的产物,经过UDP-葡萄糖焦磷酸化酶(UGP)催化后进入纤维素合成途径[30]。这个过程中UDP-葡萄糖生成UDP-葡萄糖醛酸,经由UDP-葡萄糖醛酸-差向异构酶(GAE)和半乳糖醛酸转移酶(GAUT)的催化作用,最终进入果胶的合成途径[31]。这个途径的中间产物还可作为细胞壁组分的合成前体[32]。GO 注释描述为碳水化合物代谢过程、生物合成过程等功能,是一类参与植物生长发育中能量合成消耗循环的重要基因,能量代谢的调控作用侧面影响大豆荚皮的发育,对植物的整个生长发育过程中都起着关键作用。这两个基因都可能直接或间接地参与了大豆荚皮厚的调节途径,其具体的生物学过程和分子机制还需进一步研究。
综上所述,本研究鉴定到与荚皮厚相关的重要的、可信度高的QTLqPT-n-1,预测到Gly⁃ma. 03G027100和Glyma. 03G027300是 调 控 大 豆 荚皮厚性状的重要基因。这些工作为大豆荚皮厚研究奠定了基础,为开发新的分子标记,实现大豆荚皮厚分子标记辅助育种及专化目标提供理论和材料基础。
