TLR4/NF-κB信号通路激活LncRNA RP11-20G6调控慢性阻塞性肺疾病气道炎症和重塑
2022-05-12陈训春李名兰潘碧云王燕英丁毅鹏
陈训春,李名兰,潘碧云,王燕英,丁毅鹏
慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)是一种进行性退行性肺疾病。吸烟是COPD最重要的病因,香烟烟雾中含有许多颗粒物和氧化剂,它们通过激活在小鼠肺上皮细胞和肺组织中Toll样受体(Toll-like receptor,TLR)诱导氧化应激、炎症、上皮间充质转化和纤维化[1]。Toll样受体4(Toll-like receptor 4,TLR4)是先天性免疫反应中的关键分子,它识别病原体相关分子模式(PAMPs)和损伤相关分子模式(DAMPs),并激活促炎性核因子κB(nuclear factor kappa-B,NF-κB)[2]。研究[3]表明,在COPD大鼠中,香烟烟雾通过激活肺上皮细胞和肺组织中TLR4诱导氧化应激、炎症、上皮间充质转化(EMT)和纤维化。研究[4]显示TLR4可以通过长的非编码RNA(lncRNA)调节炎症。基于以上研究,TLR4是否通过lncRNAs调节COPD炎症反应并抑制气道重塑值得关注。
LncRNAs是参与各种病理过程的调节转录本[5-6]。lncRNAs通过竞争性结合microRNAs(miRNAs)调节下游基因的表达[7]。本研究利用RNA测序法筛选了对照组、WT-COPD组和 TLR4-/- COPD组之间差异表达的LncRNAs和mRNAs。其中,lncRNA RP11-20G6.3(RP11-20G6.3)是常见的差异表达lncRNAs之一。ceRNA网络预测发现RP11-20G6.3/miR-34c-5p轴与TLR4/NF-κB通路相关。根据以往的研究[8],miR-34c-5p直接参与TLR4通路的调节,并与炎症反应密切相关。因此,笔者假设COPD中TLR4促进RP11-20G6.3的表达上调,并通过激活下游信号最终促进气道炎症和气道重塑。因此,该研究的目的是确定内源竞争RNA(competing endogenous RNAs,ceRNA)网络在COPD分子机制中的潜在作用,为COPD的治疗提供实验依据。
1 材料与方法
1.1 样本来源肺组织取自海口市人民医院20例确诊为孤立性肺肿瘤患者。将患者分为3个亚组:5名不吸烟者(非吸烟组),5名吸烟者(吸烟组),10名当前吸烟者合并COPD(COPD组)。手术切除后,取离肿瘤边缘至少5 cm的肺组织立即在液氮中冷冻,并在-80 ℃下储存,直到用于RNA分离。
1.2 质粒、试剂及仪器miR-34c-5p模拟物及其阴性对照模拟物(miR-NC),以及RP11-20G6.3序列由广州市锐博生物科技有限公司合成并克隆到pcDNA3.1(+)载体(pcDNA3.1-20G6.3)。
娇子香烟购自中国成都卷烟厂(每支卷烟含焦油11 mg、尼古丁0.9 mg);脂多糖,白细胞介素-1β(interleukin-1β,IL-1β)、白细胞介素-6(interleukin-6,IL-6)和肿瘤坏死因子(tumor necrosis factor-α,TNF-α)ELISA试剂盒购自美国Sigma-Aldrich公司;RIPA裂解缓冲液购自北京Solarbio公司;一抗(TLR4、P-IκBα、IκBα、P-NF-κB、NF-κB和GAPDH)均购自美国Abcam公司;TRIzol、LipofectamineTM3000购自美国Invitrogen公司;高容量cDNA反转录试剂盒购自美国Qiagen公司;PCR引物购自美国Thermo fisher公司;NEBNext® UltraTMII RNA文库制备试剂盒购自美国NEB公司。
7300实时PCR系统购自美国Applied Biosystem公司;Agilent 2200 TapeStation购自美国Agilent公司;2200 TapeStation和Qubit®2.0购自美国Life Technologies公司;Veritas 9100-002工具购自美国Turner BioSystems公司。
1.3 动物及COPD模型建立野生型(WT)和TLR4基因敲除(KO)C57BL/6雄性小鼠(6~8周龄)购自中国科学院上海实验动物中心,饲养在无病原体的动物房中,提供食物和水。参照文献[3]方法建立COPD模型,具体操作:小鼠被放置在一个有机玻璃室(50 cm×60 cm×70 cm),由一次性过滤器覆盖。在第1周,这些动物每天接受4支香烟,每次间隔30 min,每周5 d。从第2周开始,小鼠每天暴露于6支香烟的烟雾中,剂量维持到第6周。在第3周和第5周末,小鼠鼻腔内滴入脂多糖(Lipopolysaccharide,LPS)(30 μg/6 μl)。第2组和第4组小鼠于第3周至第6周每天腹腔注射pcDNA3.1-NC或pcDNA3.1-20G6.3(5×107IU)。最后一次暴露香烟后1周处死动物,收集支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)和肺组织。
1.4 免疫印迹分析采用RIPA裂解缓冲液从肺组织中提取蛋白质。将含有20 μg蛋白质的样品与SDS样品缓冲液混合并煮沸,然后在10%的SDS-PAGE凝胶上分离并转移到PVDF膜上。在室温下用5%脱脂牛奶封闭膜2 h,然后用相应的主要一抗:[TLR4(1 ∶4 000稀释)、P-IκBα(1 ∶1 000稀释)、IκBα(1 ∶1 000稀释)、P-NF-κB(1 ∶2 000稀释)、NF-κB(1 ∶2 000稀释)和GAPDH(1 ∶5 000稀释)]在4 ℃下培养过夜。最后用HRP结合的二抗(1 ∶5 000稀释)孵育2 h。用增强化学发光系统观察蛋白质条带。通过GAPDH免疫印迹法验证样品的等载量。用图像测量软件对谱带强度进行量化。
1.5 肺组织学评价小鼠左肺用4%甲醛固定,石蜡包埋,切片厚3~4 μm。肺切片用HE染色和Masson三色染色进行检测。炎症评分如前所述[9]。支气管周围和血管周围炎症的程度由主观评分0到3来评估。当检测不到炎症时,判定值为0;偶尔有炎性细胞缠绕时,值为1;炎性细胞包围大多数支气管或血管时,值为2;当大多数支气管或血管被一层厚的(超过5个细胞)炎性细胞包围时,值为3。全肺炎症定义为支气管周围和血管周围炎症评分的平均值。
1.6 酶联免疫吸附试验(ELISA)BALF中的IL-1β、IL-6和TNF-α水平使用ELISA试剂盒进行分析。
1.7 定量实时PCR(RT-PCR)使用TRIzol从肺组织中提取RNA。使用高容量cDNA反转录试剂盒将总RNA转换为cDNA。所有实时PCR反应均使用7300实时PCR系统进行。以GAPDH为管家基因,用比较ΔΔCT法计算mRNAs的相对表达水平。使用引物序列见表1所示。

表1 引物序列
1.8 RNA测序与功能富集分析使用TRIzol从肺组织中分离总RNA。使用Agilent 2200 TapeStation评估RNA完整性。RIN评分>7的纯化RNAs进行反向转录,然后使用NEBNext®UltraTMII RNA 文库制备试剂盒进行接头连接和低周期富集。使用安捷伦2200 TapeStation和Qubit®2.0对纯化后的文库进行评估,然后稀释至10 pmol/L,在对端流动细胞上原位生成簇,并在HiSeq3000上进行测序(2×150 bp)。清除低质量以及包含适配器和poly-N序列的读取后,获得干净的读取数据。然后使用HISAT2默认参数将读取数据与小鼠参考基因组mm10对齐,通过HTseq转换为每个基因模型的读取计数。差异表达通过DEseq评估,以读取计数为输入,进行Benjamini-Hochberg多重测试校正。差异表达基因(DEGs)以折叠变化>2和校正P<0.05为阈值进行筛选。利用RNAhybrid (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid)对ceRNA网络进行了预测。
1.9 双荧光素酶报告分析将Lnc RNA RP11-20G6.3和TLR4克隆到pisCHECK载体中,产生荧光素酶报告系统。通过LipofectamineTM3000介导的基因转移将每孔100 ng报告系统和100 nmol/L mmu-miR-34c-5p试剂转染到293 T细胞。 8 h后用完整培养基代替培养基。转染48 h后收获细胞,使用Veritas 9100-002工具测量荧光素酶活性,并标准化为海肾荧光素酶活性。
1.10 统计学处理采用SPSS 18.0软件进行统计分析,所有结果均以平均值±标准差表示。数据分析采用Studentt检验、双因素方差分析(ANOVA)和Dunnett检验。RP11-20G6.3表达与FEV1/用力肺活量的比值(forced expiratory volume in one second/forced vital capacity,FEV1%)、Col1a1的关系采用Spearman相关分析。P<0.05认为差异有统计学意义。
2 结果
2.1 TLR4缺乏减轻COPD小鼠的气道炎症和重塑为了确定TLR4在COPD中的潜在作用,首先分析了野生型(WT)COPD小鼠肺组织中的表达,并检测到TLR4蛋白(t=11.264,P<0.001)和mRNA(t=4.312,P=0.003)水平高于对照小鼠(图1A、1B)。在COPD模型中,与WT小鼠相比, TLR4-/- 小鼠肺组织炎症减弱,肺组织NF-κB相关蛋白表达降低(图1C、1D)。与这些一致, TLR4-/- 小鼠BALF中炎症因子(IL-1β、IL-6和TNF-α)水平低于WT小鼠(图1E)。观察COPD气道结构慢性重塑的特征性变化。如图2A、2B所示,WT组小鼠支气管周围三色染色(Masson's trichrome)面积大于 TLR4-/- 小鼠(t=3.910,P=0.007)。这些结果提示TLR4是引发COPD小鼠的气道炎症和重塑的重要因素。
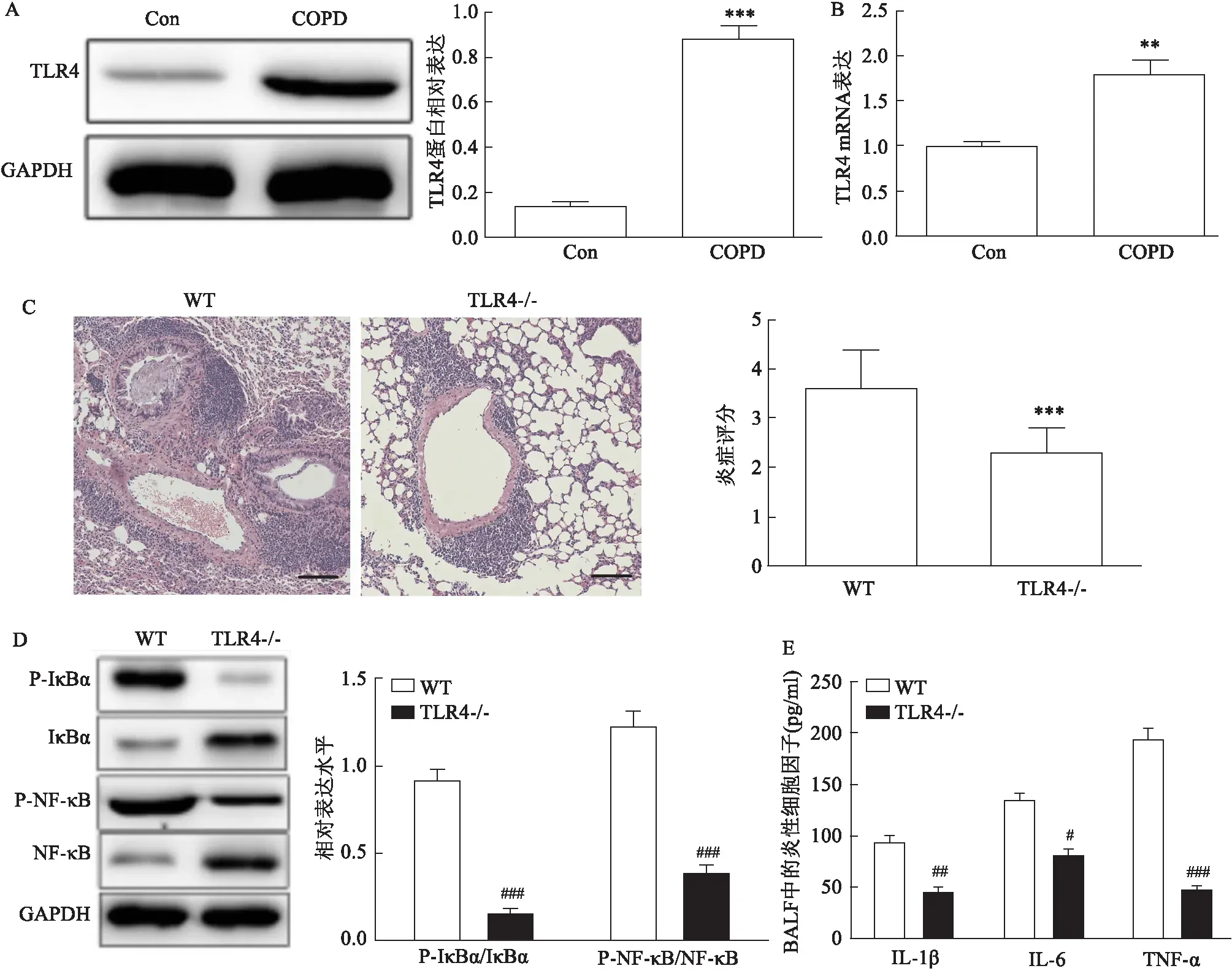
图1 TLR4缺乏减轻COPD小鼠气道炎症A:WT对照和COPD小鼠的肺组织中TLR4蛋白水平;B:WT对照和COPD小鼠的肺组织中TLR4 mRNA水平;C:WT和 TLR4-/- COPD小鼠肺组织代表性HE染色及炎症评分 ×100;D:WT和 TLR4-/- COPD小鼠肺组织NF-κB相关蛋白表达;E:WT和 TLR4-/- COPD小鼠BALF中炎症因子水平;与Con组比较:**P<0.01,***P<0.001;与WT组比较:#P<0.05,##P<0.01,###P<0.001

图2 TLR4缺乏减轻COPD小鼠气道重塑A:肺组织中Masson三色染色(黄色箭头表示胶原沉积) ×100;B:以Masson三色染色的支气管周围区域来量化皮下纤维化的程度;与WT组比较:**P<0.01
2.2 RP11-20G6.3在COPD中的异常表达WT-Con和WT-COPD小鼠的转录组分析显示621个差异表达的lncRNAs(DELs)和2 601个差异表达的基因/mRNAs(DEGs)。在这些DEGs中,328个lncRNAs上调,293个下调,而1 897个和704个mRNAs分别上调和下调。类似地,在WT-COPD和 TLR4-/- -COPD小鼠之间,124个lncRNAs和511个mRNAs上调,158个lncRNAs和899个mRNAs下调。然后预测DELs和DEGs之间的ceRNA网络,KEGG途径分析表明NF-κB途径和TLR途径富集(图3A)。RP11-20G6.3是常见的差异表达lncRNAs之一;进一步RT-qPCR验证表明,RP11-20G6.3是TLR4调节动物中差异表达最大的LncRNA(图3B)。为了确定RP11-20G6.3表达是否与COPD相关,并确定其与气道重塑的可能相关性,进行了RT-qPCR。RP11-20G6.3在COPD患者的肺组织中上调,并与FEV1%相关(ρ=0.549,P=0.047)(图4A、4B)。这些结果表明,随着COPD的进展,RP11-20G6.3水平升高,其在病变组织中的上调依赖于TLR4。
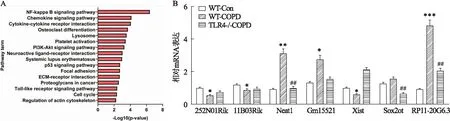
图3 COPD后RP11-20G6.3上调,与疾病严重程度相关A:KEGG通路分析WT-Con、WT-COPD和 TLR4-/- COPD小鼠的DELs和DEGs;B:WT-Con、WT-COPD和 TLR4-/- COPD小鼠肺组织的LncRNA水平;与WT-Con组比较:*P<0.05,**P<0.01,***P<0.001;与WT-COPD组比较:##P<0.01

图4 RP11-20G6.3在COPD患者中表达较高A:qRT-PCR检测非吸烟者(n=5)、吸烟者(n=5)和COPD患者(n=10)肺组织中RP11-20G6.3的表达;B:RP11-20G6.3表达与FEV1%的Spearman相关分析;与非吸烟者组比较:*P<0.05,**P<0.01
2.3 RP11-20G6.3逆转TLR4缺乏减轻COPD小鼠气道炎症和重塑为了确定RP11-20G6.3在气道重塑发病机制中的潜在作用,将RP11-20G6.3质粒滴鼻给于 TLR4-/- -COPD小鼠。与其他组相比,加入RP11-20G6.3质粒增加了肺组织RP11-20G6.3水平(图5A)。HE染色显示,RP11-20G6.3质粒干预后 TLR4-/- 小鼠肺组织炎症增强(t=3.952,P=0.007),支气管周围Masson三色面积增加(t=4.062,P=0.004),并且BALF中炎症因子(IL-1β、IL-6和TNF-α)水平提高(图5B-5D)。因此,RP11-20G6.3介导TLR4诱导的COPD小鼠气道炎症和重塑。

图5 RP11-20G6.3逆转TLR4缺乏减轻COPD小鼠气道炎症和重塑A:qRT-PCR检测小鼠肺组织中RP11-20G6.3的表达;与WT组比较:△△P<0.01;与 TLR4-/- 组比较:###P<0.001;B:经RP11-20G6.3处理的 TLR4-/- COPD小鼠肺组织代表性HE染色及炎症评分 ×100;C:经RP11-20G6.3处理的 TLR4-/- COPD小鼠肺组织代表性Masson三色染色及纤维化的程度(黄色箭头表示胶原沉积) ×100;D:经RP11-20G6.3处理的 TLR4-/- COPD小鼠BALF中炎症因子水平;a:WT组;b:TLR4-/-组;c:TLR4-/-+pcDNA3.1-NC组;d:TLR4-/-+pcDNA3.1-20G6.3组;与 TLR4-/- +pcDNA3.1-NC组比较:**P<0.01,***P<0.001
2.4 RP11-20G6.3海绵miR-34c-5p上调Col1a1使用RNAhybrid预测了靶向RP11-20G6.3的ceRNA网络,并发现RP11-20G6.3海绵miR-34c-5p上调Col1a1。使用在线生物信息数据库预测miRNA靶点,并确定miR-34c-5p在RP11-20G6.3 mRNA中具有相关的结合位点(图6A)。miR-34c-5p与RP11-20G6.3之间的关系通过荧光素酶报告基因检测进行验证。如图6B所示,miR-34c-5p抑制WT控制的荧光素酶报告活性(t=7.837,P<0.001),但不抑制突变型RP11-20G6.3启动子的活性。miR-34c-5p与Col1a1 mRNA的3UTR相互作用(图6C)。miR-34c-5p还抑制Col1a1启动子报告基因的荧光素酶活性(t=4.124,P=0.002)(图6D)。因此,RP11-20G6.3和Col1a1的3'-UTR序列是miR-34c-5p的直接靶点。此外,COPD患者的Col1a1水平升高,并与RP11-20G6.3呈正相关(ρ=0.936,P<0.001)(图6E、6F)。总之,RP11-20G6.3在COPD中充当miR-34c-5p的ceRNA,并通过海绵作用使后者上调Col1a1。
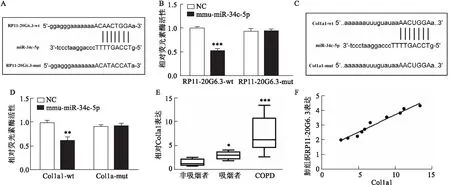
图6 RP11-20G6.3海绵miR-34c-5p上调COPD后Col1a1A、B:RP11-20G6.3 mRNA的miR-34c-5p结合序列(A)及测定荧光素酶活性(B);C、D:Col1a1 mRNA的miR-34c-5p结合序列(C)及测定荧光素酶活性(D);E:qRT-PCR检测非吸烟者(n=5)、吸烟者(n=5)和COPD患者(n=10)肺组织中Col1a1的表达;F:RP11-20G6.3表达与Col1a1的Spearman相关分析;与NC或非吸烟者组比较:*P<0.05,**P<0.01,***P<0.001
3 讨论
COPD因其复杂的病理机制,目前尚无有效的治疗方法。气道炎症和重塑是COPD呼吸系统损伤的重要因素[10]。目前认为气道壁损伤和修复的重复循环引起的慢性炎症最终导致气道重塑[11]。本研究显示,TLR4在COPD中上调,其缺乏可通过下调肺组织NF-κB相关蛋白表达来减轻COPD小鼠BALF中炎症因子释放,并减轻气道纤维化的程度。这些结果与先前的研究[2]结果一致,即TLR4的下调可以减轻炎症并阻止COPD后的气道重塑。NF-κB家族是研究最广泛的转录因子,因为它在炎症相关反应中起关键作用[12]。它们可以通过N端DNA结合域形成同源二聚体和异二聚体,并与κB位点的各种相关靶DNA序列结合以调节基因表达。TLR4是先天性免疫反应中的一个关键分子,它识别PAMPs(主要是LPS),激活NF-κB途径,这可能是其参与COPD炎症和气道重塑的最重要信号[2]。
LncRNAs调节各种病理状态下的炎症反应,其异常表达是多种肺部疾病的分子标志,提示lncRNAs在这些肺部疾病的发病机制中具有潜在的作用[13]。研究[14]表明,lncRNA-NEAT1在肺炎中通过海绵miR-193a-3p介导LPS诱导的细胞凋亡和炎症损伤。先前的研究[15]表明,差异表达的lncRNAs和miRNAs之间的相互作用可能为确定COPD全新的治疗靶点提供了新的思路。此外,最近研究[16]显示,NNT-AS1基因敲除可阻断香烟烟雾提取物对细胞增殖、凋亡、炎症和气道重塑的抑制作用。与这些发现一致,RP11-20G6.3在COPD患者和WT-COPD小鼠肺组织中异常上调,并与COPD患者的FEV1%相关,而强制表达RP11-20G6.3则加重了TLR4缺失的药理抑制作用。因此,RP11-20G6.3是TLR4诱发COPD气道炎症、重塑的重要介质。
大量证据支持LncRNAs通过多种机制靶向miRNAs间接调控基因表达,从而改变了多种生物过程[6]。它们可以充当miRNA海绵(也称为ceRNAs),与miRNAs竞争靶mRNAs,被加工成miRNA,并将miRNA转运到靶mRNAs。在COPD中发现了多种ceRNAs。例如,Mei et al[16]报道了lncRNA-NNT-AS1通过靶向miR-582-5p来促进香烟烟雾提取物对细胞增殖、凋亡、炎症和气道重塑的作用。同样,本研究表明RP11-20G6.3/miR-34c-5p/Col1a1 ceRNA网络调节COPD气道重塑。胶原蛋白是一种主要的细胞外基质蛋白,而I型胶原尤其在炎症中起着信号分子的作用。Zhang et al[17]研究发现,I型胶原通过增加ROS水平刺激小鼠腹腔巨噬细胞的募集和聚集,以及促炎性细胞因子的产生。Molokanova et al[18]发现,I型胶原缺乏减弱受损肝脏的炎性细胞活化/募集。本研究在COPD患者的肺组织标本中检测到较高的Col1a1水平,进一步强调了其作为RP11-20G6.3下游信号与COPD气道炎症和重塑的关系。
总之,TLR4/NF-κB将COPD损伤信号传递并激活下游RP11-20G6.3/miR-34c-5p/ Col1a1的ceRNA网络触发气道炎症、重塑。
