肿瘤转移相关基因1短式在大肠杆菌中的表达*
2022-05-10王攀琦刘星吟魏瑶璐宁志丰沈定文
刘 洋,王攀琦,王 芳,刘星吟,董 兰,魏瑶璐,宁志丰,沈定文
(1.湖北科技学院医学部药学院,湖北 咸宁 437100;2.湖北科技学院医学部基础医学院)
肿瘤的复发和转移是患者长期生存的最大威胁,也是其主要死因[1]。转移作为癌症的重要标志之一,转移相关蛋白(metastasis associated proteins,MTA)是与肿瘤转移密切相关的一小类转录辅助调节因子,这些蛋白质是核重塑和去乙酰化复合体(nucleosomeremodeling deacetylase,NuRD)的组成部分[2-3]。在这其中肿瘤转移相关基因1(MTA1)在肿瘤的转移中发挥重要作用,与此同时,肿瘤转移相关基因1短式(MTA1s)是一种自然发生选择性剪接的MTA1变体[4],并且与肿瘤转移有所关联,MTA1s是由外显子14中一个隐性剪接位点的选择性剪接、47个碱基对核苷酸的缺失和一个添加新的33个氨基酸的移码而产生的。有研究发现[5-6]MTA1s在乳腺癌MCF-7细胞中可作为雌激素受体(ER)的抑制物,阻止了MCF-7细胞过度表达ER,由此证实MTA1s的抗雌激素活性凸显了其治疗潜力。随着靶向治疗及基因治疗的不断发展,靶向单克隆抗体药物针对肿瘤的治疗较为显著[7],抗肿瘤抗体药物已经成为肿瘤领域不可或缺的治疗手段。近年来,肿瘤免疫治疗尤为突出,具有免疫调节的抗体药物,或能增加肿瘤抗原性,或能促进肿瘤T细胞浸润,已经取得了一定的临床效益[8]。经原核表达分离且纯化的重组蛋白对后续研究单克隆抗体和多克隆抗体较为关键。本实验采用PCR扩增人MTA1s的蛋白编码区、构建原核表达载体,并用IPTG诱导MTA1s重组蛋白在大肠杆菌BL21中进行表达,为后续的相关研究奠定基础。
1 材料与方法
1.1 实验材料
1.1.1 细胞株与主要仪器
人乳腺癌MCF-7细胞购自中国典藏物培养中心;纯水机(上海和泰);超净工作台(苏州安泰空气技术公司);恒温水浴锅(江苏金坛市中大仪器厂);水平摇床(上海琪特);低温离心机(Eppendorf);细胞超声破碎仪(JY92-IIPN宁波新芝生物科技股份有限公司);琼脂糖凝胶电泳仪;微型垂直电泳仪(Tanono);PCR仪(LifeEco);气浴恒温振荡器(江苏金大坛中大仪器厂);超低温冰箱(海尔)。
1.1.2 主要试剂
HindⅢ、EcoRⅠ限制性内切酶购自NEB公司;phanta Max Super Fidelity DNA Polymerase、ClonExpressⅡ One Step Cloning Kit购自诺唯赞公司;胎牛血清购自杭州四季青公司;DMEM培养基购自Gibco公司;pET-28a购自淼灵质粒平台;酵母粉、蛋白胨、总RNA提取试剂盒、IPTG溶液(50mg/mL)、卡那霉素溶液(100mg/mL)购自Solarbio公司;Nacl购自上海源叶生物公司;TAE(50X)、核酸染料购自biosharp公司;cDNA第一链合成试剂盒、考马斯亮蓝快速染色液、脱色液购自碧云天生物公司;Gene JET Gel Extraction Kit购自therno scientific公司;Super DNA Marker、2×Super Pfx MasterMix、EndoFree Plasmid Midi Kit购自康为世纪公司;BL21(DE3)感受态细胞、DH5α感受态细胞购自昂羽生物公司。
1.2 实验方法
1.2.1 细胞培养
人乳腺癌细胞MCF-7置于含10% 胎牛血清的DMEM培养液在37℃、5% CO2培养箱中培养。
1.2.2 总RNA提取与反转录cDNA
取对数生长期的MCF-7细胞,弃培养液,用无菌的磷酸盐缓冲液(phosphate buffer saline,PBS)清洗贴壁细胞3次,取裂解液1mL按说明书提取细胞内总RNA,对提取的总RNA进行浓度测定后,取3μg作为合成cDNA第一链的模板,还需添加引物oligo(dT)18 Primer(0.5μg/μL)1μL,DEPC-treated water补加到12μL,70℃孵育5min,冰浴冷却,4℃微离心后加入Reaction Buffer(5X)4μL,RNase Inhibitor(20U/μL)1μL,dNTP Mix(10mM each)2μL,42℃孵育60min,70℃ 10min终止反转录反应后即得到cDNA。
1.2.3 引物设计及目的片段的扩增
采用同源重组的方法进行引物设计,在插入片段正反向扩增引物的5′端引入线性化载体两末端同源序列,使扩增后的插入片段5′和3′最末端分别带有和线性化克隆载体两末端对应一致的同源序列且不包含酶切位点。在本实验中,载体选用pET-28a,按NCBI GenBank MTA1s基因序列(NM-001203258.2)设计引物,分析后选择HindⅢ和EcoRⅠ作为酶切位点。引物设计如下:
MTA1s-Forward:5′-3′ggatccgcgacccagaattcatggccgccaacatgtacag MTA1s-Reverse:5′-3′tcgagtgcggccgcaagcttttactgcggtttggtcctgg PCR反应体系(50μL):MTA1s-Forward,MTA1s-Reverse各2.5μL,模板1μL,2×Phaha Max Buffer 25μL,dNTP Mix 1μL,Phaha Max Super-Fidelity DNA polymerase 1μL,ddH2O 17μL。按95℃预变性3min,95℃变性15s,67℃退火15s,72℃延伸1min30s,循环35次,72℃ 5 min的反应条件扩增MTA1s基因中的蛋白编码区DNA片段。反应结束后使用1.0%琼脂糖凝胶进行DNA电泳分析,并将目的条带切胶回收,放-20℃保存备用。
1.2.4 原核表达载体的构建
用HindⅢ和EcoRⅠ限制性内切酶将pET-28a载体线性化,双酶切反应体系1(50μL)为:EcoRⅠ 2μL,HindⅢ 2μL,NE BufferTM2.1 5μL,pET-28a 10μL,ddH2O 31μL,37℃ 75min。反应结束后用1.0%琼脂糖凝胶进行电泳分析是否酶切完全,并将线性化载体切胶回收,放-20℃保存备用。采用一步克隆试剂盒进行重组反应,反应体系为:线性化载体2μL,插入片段2μL,5×CE Ⅱ Buffer 4μL,ExnaseⅡ 2μL,ddH2O 10μL,37℃反应30min后,将重组产物转入DH5α感受态细胞,涂LB固体平板,37℃孵育过夜,次日挑取单克隆,摇菌,部分做菌液PCR及测序(擎科生物科技有限公司)鉴定。剩余部分菌液与50%甘油按1∶1冻存于-80℃。
1.2.5 重组质粒(pET-28a-MTA1s)的提取及双酶切验证
经测序鉴定和菌液PCR鉴定正确后,将冻存于-80℃的菌液活化,1∶1 000添加质粒上所带抗性的抗生素,先按1∶10在37℃摇床摇菌1h,再按1∶100的比例37℃振摇过夜,第2d将培养过夜的菌液提取质粒,经Nanodrop测定质粒浓度后,放于-20℃保存。将提取的重组质粒(pET28a-MTA1s) 用HindⅢ和EcoRⅠ限制性内切酶双酶切,双酶切反应体系2(50μL)为:EcoRⅠ 2μL,HindⅢ 2μL,NE BufferTM2.1 5μL,pET-28a-MTA1s 4μg,ddH2O 33μL 37℃反应75min。反应结束后使用1.0%琼脂糖凝胶进行DNA电泳分析。
1.2.6 重组蛋白的诱导表达
为研究pET-28a-MTA1s原核表达载体能否成功诱导表达目的蛋白,我们将重组质粒pET-28a-MTA1s转入BL21(DE3)感受态细胞,并涂带有相应抗性的LB固体培养板,37℃孵育过夜,次日,挑取单克隆于灭菌的LB液体培养基中(按1∶1 000的比例添加卡那霉素),37℃培养4h左右并按菌液与甘油1∶1的比例冻存于-80℃,次日,取冻存菌液按1∶100比例接种于带相应抗性的灭菌LB液体培养基中并在37℃恒温摇床活化12h后继续按1∶100扩大培养,继续培养3~4h左右,当菌体生长达到对数期时先取出2mL菌液作为对照,剩余菌液加入终浓度为0.5mmol/L异丙基硫代半乳糖苷(Isopropyl β-D-Thiogalactoside,IPTG)诱导表达,诱导温度为16℃,诱导时间为12h。
1.2.7 裂解重组蛋白并于SDS-PAGE电泳验证
诱导完后,取2mL菌液标记为加IPTG后沉淀,离心,加入蛋白上样缓冲液后,98℃ 10min用于电泳。剩余诱导的菌液离心收集诱导后的菌体用PBS重悬菌体沉淀置于冰上超声破碎,当上清明显变为清亮,即为破碎完全,12 000rpm,离心20min,分别收集上清和沉淀,分别取破碎后的蛋白上清液和破碎后沉淀加入蛋白上样缓冲液,98℃金属浴加热10min使蛋白变性后,分别吸取10μL上清液和沉淀进行SDS-PAGE电泳,电泳结束后,立即用考马斯亮蓝染色液染SDS-PAGE胶4~5h,脱色后拍照观察蛋白诱导表达情况。
1.2.8 蛋白免疫印迹鉴定重组质粒
将制备的样品经SDS-PAGE电泳后,采用湿转的方法,用1X转膜缓冲液冰浴条件下275mA转印90min,转印结束后,将PVDF膜取出,将其在室温条件下用5%的脱脂牛奶进行封闭1h后,TBST洗膜3次,每次10min,将His-Tag Mouse Monoclonal Antibody用一抗稀释液按1∶5 000稀释,将PVDF膜在4℃摇床孵育过夜。次日,一抗孵育结束后,用TBST清洗3次,10min/次,用TBST将Goat-Anti-Mouse IgG/HRP按说明书比例稀释并在室温条件下将PVDF膜孵育1h,TBST清洗3次,10min/次,充分洗膜,将膜上滴加ECL化学发光液。孵育2~3mim后,在Image Quant LAS500上进行显影操作。
2 结 果
2.1 PCR扩增目的片段
以MCF-7细胞为模板进行PCR扩增目的条带,将扩增产物用1.0%琼脂糖凝胶进行DNA电泳验证,如图1所示,目的片段为1 293bp,扩增出大小正确的DNA片段,符合预期结果并将条带切胶回收。
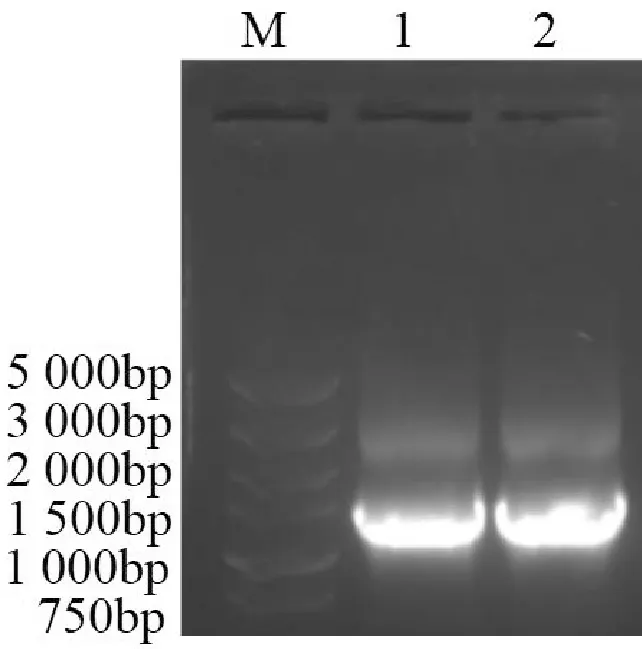
M:DL5 000 DNA Marker;1-2:MTA1s基因的PCR扩增结果
2.2 pET-28a质粒双酶切
以HindⅢ和EcoRⅠ限制性内切酶对pET-28a载体线性化,如图2所示,1代表空质粒,即未进行酶切的质粒,琼脂糖凝胶电泳显示为两条目的带,而酶切完全的线性化载体仅为一条带,符合实验预期,并将线性化载体切胶回收。
M:Super DNA Marker;1:pET-28a空质粒;2-4:pET-28a线性化质粒
2.3 pET-28a-MTA1s重组质粒菌液PCR及测序鉴定
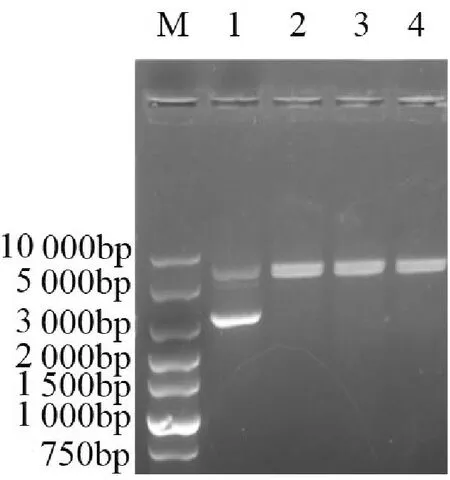
将线性化载体与经过胶回收得到的目的片段进行克隆(重组反应)后,将重组产物转化入DH5α感受态细胞中,涂板,挑单菌落,摇菌4h左右取部分菌液送测序公司进行鉴定。NCBI中MTA1s基因序列(NM-001 203 258.2)与测序结果进行比对,如图3所示,测序结果正确且无突变,表明重组载体构建成功。并以菌液为模板进行菌液PCR验证,PCR反应结束后,将反应产物用1.0%琼脂糖凝胶进行DNA电泳分析,如图4所示,以菌液为模板扩增出的特异性条带与胶回收后的目的条带位置一致。
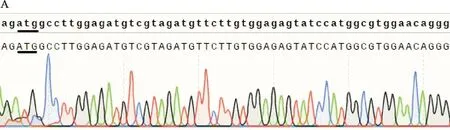
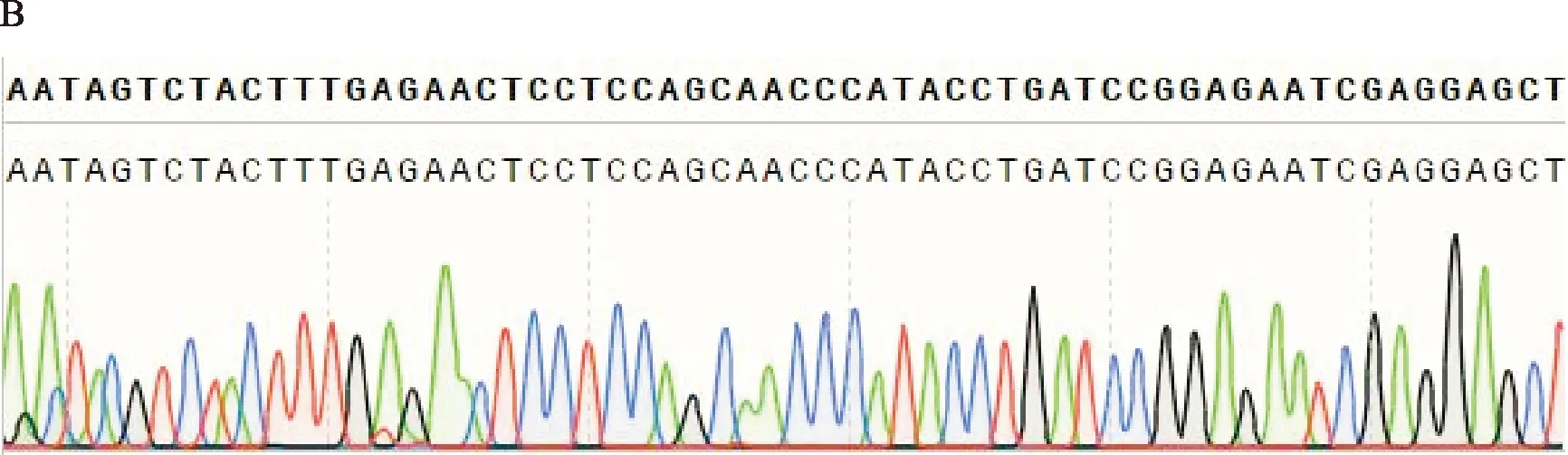
A:pET-28a-MTA1s Forward;B:pET-28a-MTA1s Reverse
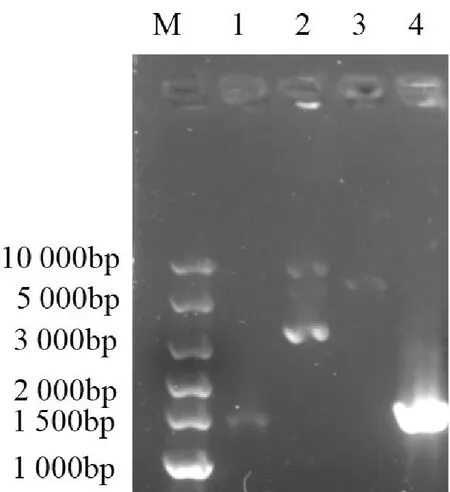
M:Super DNA Marker;1:胶回收后MTA1s目的片段(1 293bp);2:pET-28a空质粒;3:胶回收后pET-28a线性化载体;4:菌液PCR扩增得到的MTA1s目的片段(1 293bp)
2.4 重组质粒pET-28a-MTA1s双酶切鉴定
将阳性转化子过夜摇菌提取重组质粒,按双酶切反应体系2对pET-28a-MTA1s和空质粒pET-28a进行酶切鉴定。结果如图5所示,重组质粒pET-28a-MTA1s双酶切后出现2条目的条带而空质粒pET-28a酶切完全后仅有一条目的带,与预期结果一致。此外,基因测序结果也表明插入的目的片段与已知序列一致,未发生突变。以上结果表明重组质粒pET-28a-MTA1s构建成功,MTA1s目的片段已插入表达载体中。
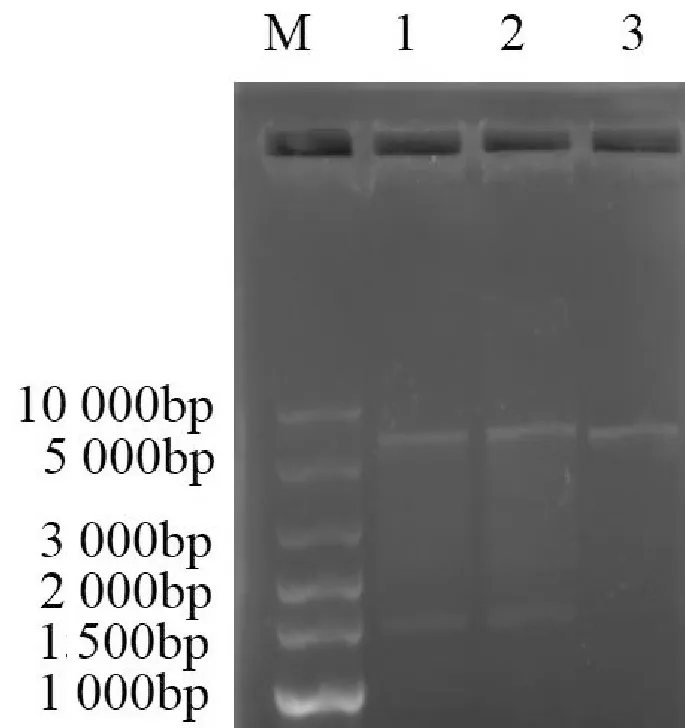
M:Super DNA Marker;1-2:双酶切后的pET-28a-MTA1s载体;3:双酶切后的pET-28a空质粒
2.5 MTA1s重组蛋白SDS-PAGE鉴定表达情况
将测序正确的质粒pET-28a-MTA1s转入BL21(DE3)感受态细胞中,并将重组菌37℃培养至OD600nm值为0.6~0.8时,取出部分菌液作为对照,剩余部分加入IPTG(0.5mmol/L)低温诱导过夜后收集菌体,冰浴条件下超声破碎细胞20min后,低温条件下12 000转离心20min,分别收集上清和沉淀,经SDS-PAGE检测重组菌诱导后产物与重组菌诱导前产物。如图6所示,超声破碎离心后,目的条带只出现在重组菌诱导产物的沉淀中,表明重组蛋白MTA1s在BL21中成功表达,主要以包涵体形式存在。
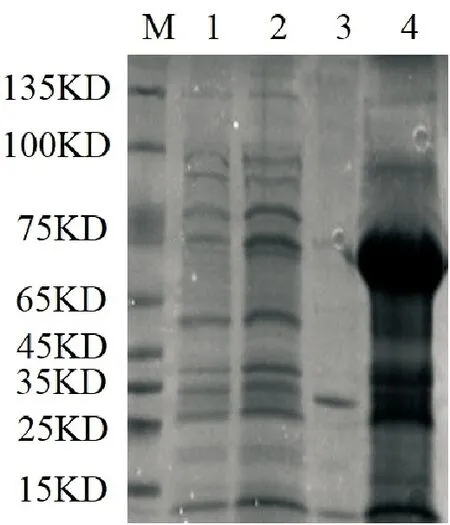
M:protein marker;1:重组菌诱导前产物;2:重组菌诱导后产物;3:重组菌诱导产物的上清;4:重组菌诱导产物的沉淀
2.6 Western blot鉴定重组载体表达情况
由于pET-28a-MTA1s重组质粒带有his标签,将未加IPTG诱导的菌液和加入IPTG诱导的菌液收菌后将菌体超声破碎的上清和沉淀分别一起孵育his抗体进行Western blot鉴定。目的片段为1293bp,其对应的分子量50~60KD。如图7所示,重组质粒pET-28a-MTA1s被IPTG诱导后沉淀有一条明显的蛋白条带,大小与目的蛋白一致。表明构建的重组载体可以正确进行目标蛋白的表达。
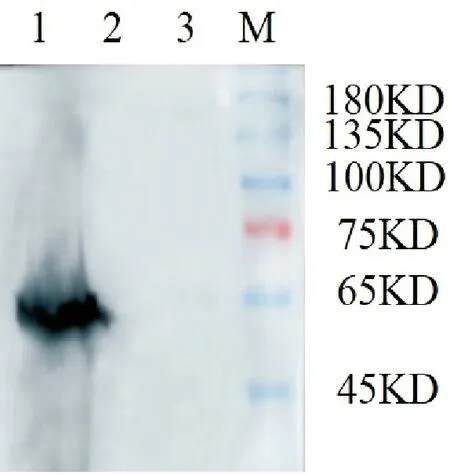
M:protein marker;1:将重组质粒pET-28a-MTA1s被IPTG诱导后沉淀;2:将重组质粒pET-28a-MTA1s被IPTG诱导后上清;3:重组质粒pET-28a-MTA1s诱导前产物
3 讨 论
MTA家族由三个基因构成,包括MTA1、MTA2和MTA3以及一些类似分子MTA1s、MTA1ZG29p和MTA3L[9]。这些蛋白作为核重塑和去乙酰化复合物(nucleosomeremodeling deacetylase,NuRD)的组成部分,通过改变靶基因染色质上的乙酰化和甲基化等表观遗传来调节基因的表达[10],此外,MTA家族也通过调节细胞骨架、雌激素信号通路等途径促进肿瘤的侵袭和转移[11]。相关文献表明[12-16],MTA1在多种癌症中过表达并促进肿瘤的侵袭与转移,包括乳腺癌、胃癌、肺癌、肝癌、白血病等。MTA家族与人类癌症密切相关,并且还发现了两种MTA1变体即ZG29p和MTA1s,ZG29p是MTA1的N端截断形式,而MTA1s是MTA1的C端截断形式[17]。然而作为MTA1的剪切类似分子MTA1s在对肿瘤的进展中表现出相反的作用,MTA1s是由MTA1基因第14个外显子处选择性断裂、在1 224~1 272bp处缺失47个碱基,由此而产生的MTA1s蛋白含有一新的由33个氨基酸组成、带有核受体结合序列即亮氨酸-精氨酸-异亮氨酸-亮氨酸-亮氨酸(Leu-Arg-Ile-LeuLeu,LXILL)的结构,且定位于乳腺癌细胞的细胞质中[18],该结构对于存在于胞质中封闭雌激素受体的作用相关[19]。Kumar等人[20]发现MTA1s具有抗雌激素(ER)的作用,其C端特有的结构使其定位于乳腺癌细胞的胞质中,通过对胞质中的ER进行封闭,阻止了核信号的传递使得ER的转录受到抑制。Singh等[21]通过一系列研究表明在MCF-7乳腺癌细胞中,MTA1s肽能够有效抑制ER,并且还发现MTA1s的抗雌激素效应是由于它能够与雌激素受体的下游激活因子相竞争而发挥作用,此外,MTA1s肽可有效抑制雌激素诱导的MCF-7细胞增殖同时也可阻断MCF-7细胞过度表达雌激素受体从而减缓肿瘤的发生发展[22]。Wang等[23]也表明MTA1s肽的结构特点和抗雌激素活性凸显了其治疗潜力。因此,有必要单独研究MTA1s的生物学功能。首先需要获得的是MTA1s蛋白,本文通过构建重组原核表达载体pET-28a-MTA1s,将其在大肠杆菌BL21中进行表达,从考染结果得出重组蛋白在沉淀中表达较为明显,为后续研究MTA1s的生物学功能奠定基础。
本实验利用同源重组的方法来设计引物并构建重组载体,与常规的双酶切构建载体方法相比相对简便,也可提高连接的效率[24-25]。二者的差别主要在引物设计上。设计引物时上游引物5′端前加上EcoRⅠ酶切位点及该酶切位点在pET-28a-MTA1s载体中对应前面14个碱基载体片段,下游引物5′端前面加的是HindⅢ酶切位点及该酶切位点在pET-28a-MTA1s载体中对应后面的14个碱基载体片段,此方法设计引物有利于PCR扩增得到目的片段后的重组反应[26-27]。
迄今为止,肿瘤的治疗仍面临巨大挑战,与传统放化疗相比,抗体药物具有高靶向性、毒副作用小和疗效好等特点,已成为肿瘤治疗的重要辅助手段[28]。此外,单克隆抗体从单纯的科研工具逐渐演变成生物药物,用于自身免疫、肿瘤和感染等疾病的治疗。Kaplon等[29]详细介绍了抗体药物的最新进展,其中,值得注意的是,有44种抗体疗法正处于癌症晚期临床试验中。在肿瘤微环境中尤其是在乳腺癌中MTA1s通过将受体隔离在细胞质中,阻止了雌激素受体信号转导,发挥其抗雌激素活性,在一定程度上能够抑制乳腺癌细胞的生长[30],在乳腺癌中发挥治疗潜力。本实验构建的pET-28a-MTA1s表达载体能够诱导表达MTA1s融合蛋白,为后续作为抗原应用或制备MTA1s单克隆抗体奠定了实验基础。
