催化裂化柴油加氢裂化生产轻质芳烃研究进展
2022-04-28高子祺李佳鑫王莉娴袁桂梅陈胜利
南 毅,高子祺,李佳鑫,王 楠,王莉娴,吴 韬*,袁桂梅,陈胜利
(1.北京石油化工学院新材料与化工学院,恩泽生物质精细化工北京市重点实验室,北京 102617;2.中国石油大学(北京)重质油国家重点实验室,北京 102249)
轻质芳烃如苯(B)、甲苯(T)和二甲苯(X)广泛用于制备塑料、橡胶和化纤等领域,是重要的有机化工原料[1]。长期以来,我国轻质芳烃一直供不应求,特别是对二甲苯,对外依存度已超过50%[2]。另一方面,催化裂化轻循环油(LCO),又称催化裂化柴油,具有芳香烃含量高和十六烷值低等特点,需与直馏柴油调和或经过适当的精制后才能作为柴油使用,或用作燃料。目前,我国催化裂化装置年加工量已达1.0×108t,LCO年产量超过1.0×107t[3]。因此,在当前柴油严重过剩且环保要求日益提高的大背景下,将具有富含芳烃和价格低廉的LCO转化为轻质芳烃,是目前解决柴油过剩和轻质芳烃短缺较为理想的途径。本文综述LCO加氢裂化生产轻质芳烃相关技术、反应规律及催化剂的研究进展。
1 LCO制取轻质芳烃技术
目前国内外已有将LCO中的芳烃(特别是双环芳烃和单环芳烃)通过加氢裂化反应转化为轻质芳烃相关的技术。
1.1 LCO-X技术
UOP公司在2007年开发了以LCO为原料生产轻质芳烃的LCO-X技术[4],其工艺流程如图1所示。该技术的核心是加氢裂化与催化重整,LCO首先经过加氢处理脱除杂质,再进行加氢裂化反应使稠环芳烃转化为轻质芳烃或环烷烃,最后通过催化重整工艺将潜芳烃(主要是环烷烃)转化为芳烃,从而取得较高的轻质芳烃收率,主要产物为二甲苯(收率约32%)和苯(收率约12%),甲苯和乙苯含量较低。

图1 LCO-X工艺流程图[4]
1.2 ARO技术
ARO(Aromatic Ring-Opening)技术是由NOVA化学品公司开发,可将LCO转化为BTX含量高的汽油[5]。该工艺由加氢处理和加氢裂化两个过程组成,LCO首先经过加氢处理一方面降低硫和氮含量,另一方面使其稠环芳烃发生选择性加氢反应,该过程使用的催化剂为NiMo/Al2O3或NiW/Al2O3;再进行加氢裂化反应得到C2~C4轻烃(收率约32%)、C5~C12烷烃(收率约48%)和C6~C9芳烃(收率约19%),该过程使用的催化剂为负载贵金属Pd分子筛[6]。
1.3 FD2G技术
FD2G技术是通过对工艺技术与催化剂的组合优化实现LCO选择性加氢,将LCO中的稠环芳烃有效地转化为轻质芳烃。产物中汽油馏分的质量分数为30%~50%。而汽油馏分中C6~C9芳烃含量达50%,其中BTX含量可达32%[7]。
1.4 RLG技术
RLG技术是以芳烃含量高的LCO为原料,采用单段串联和轻柴油部分循环的新型工艺流程,生产高辛烷值汽油调合组分或BTX[8]。该技术是将原料先加氢精制(RN-411加氢精制催化剂),再加氢裂化(RHC-100加氢裂化催化剂)。加氢裂化产物蒸馏后分出的石脑油馏分再进入铂重整装置生产低碳芳烃BTX,也可以直接作为汽油组分。在温度(380~420) ℃和氢分压(4.9~6.0) MPa条件下,通过控制LCO中芳烃的加氢转化路径,可以生产收率30%~50%和硫含量低的高辛烷值汽油组分,同时生产的柴油馏分收率为30%~40%,且十六烷值明显提高。
就现有技术而言,除LCO-X外,其他工艺均是以生成高十六烷值柴油或高辛烷值汽油为目的,在柴油过剩的当下经济效益十分有限。而LCO-X不能简单高效地将LCO转化为轻质芳烃,究其原因是LCO组成过于复杂,导致其在加氢裂化条件下,发生的反应众多,很难从调节温度、压力和氢油比等工艺条件控制反应进程来获得高BTX收率。因此,从化学反应角度入手,研究芳烃加氢裂化反应规律,开发具有针对性的高效加氢裂化催化剂,实现LCO到轻质芳烃的转化变得尤为重要。
2 LCO加氢裂化反应规律
加氢裂化反应可以看作氢气存在下的加氢反应与裂化反应的耦合。LCO中的芳烃组分包括单环芳烃和双环芳烃,及少量的三环芳烃,均可以通过加氢裂化反应转化为BTX。
2.1 单环芳烃
2.1.1 茚类、茚满和四氢萘类
Sato K等[9]在反应温度350 ℃和压力6.1 MPa条件下,以NiW/Z(Z=USY、HY、丝光沸石)为催化剂(NiO质量分数3.5%,WO3质量分数24.0%),催化四氢萘加氢裂化反应。结果表明,在该条件下四氢萘的加氢裂化反应共有3个反应方向:(1) 完全加氢得到十氢萘,再异构、开环、裂解;(2) 直接异构、开环得到丁苯或异丁苯;(3) 脱氢生成萘。四氢萘加氢裂化反应机理如图2所示。

图2 四氢萘加氢裂化反应机理[9]
Wang Q等[10]以Pt/USY为催化剂研究了反应温度(220~300) ℃、压力(2.0~6.0) MPa、氢油体积比500~750和空速(1~2) h-1对四氢萘加氢裂化反应的影响。研究表明,四氢萘在该条件下主要发生两个反应路径:四氢萘异构化生成茚满,再开环裂解生成BTX等烷基苯;四氢萘加氢饱和后再异构化,开环裂解生成小分子烷烃。Liu Y等[11]研究了分子筛类型(Beta、Y、ZSM-5)对四氢萘加氢裂化反应的影响,发现在反应温度400 ℃、氢油体积比400、压力4.0 MPa和空速(2~4) h-1条件下,三组催化剂上均不发生四氢萘完全加氢的反应。ZSM-5分子筛对异构化产物(甲基茚满)选择性最高,而Beta分子筛由于适宜的酸性和孔道结构对BTX选择性最高。
2.1.2 长链或多侧链烷基苯

2.2 双环芳烃加氢裂化反应
目前对于双环芳烃加氢裂化反应的研究较多,主要以1-甲基萘(1-MN)或萘等模型分子展开。Miki Y等[13]以NiMo/α-Al2O3为催化剂,在反应压力6.0 MPa和反应温度(390~450) ℃条件下,研究了1-MN和2-甲基萘加氢裂化反应,结果表明,双环芳烃加氢裂化反应是个复杂的平行顺序反应,反应产物多达100多种,主要有以下6类:(1) 异构化产物;(2) 部分加氢产物:甲基四氢萘及其异构体;(3) 完全加氢产物:甲基十氢萘及其异构体;(4) 开环断侧链产物:烷基苯;(5) 仅开环产物:C11烷基苯;(6) 异构未开环产物:茚满及烷基取代的茚满。结果表明,1-MN在加氢生成甲基四氢萘后仍有可能加氢生成甲基十氢萘。Kim E S等[14]以NiW/Beta为催化剂,在温度380 ℃和压力6.0 MPa条件下,进行LCO模型油(15%菲和85%1-MN的混合物)加氢裂化反应。结果表明,反应路径有两条:第一,1-MN先发生单个苯环的加氢生成甲基四氢萘,再发生脂环的异构化生成二氢茚,最后开环裂解生成轻质芳烃;第二,1-MN先发生脱烷基反应生成萘,再加氢生成四氢萘,并进一步异构化、开环裂解生成轻质芳烃。结果表明,甲基四氢萘的选择性远远高于萘,说明1-MN更倾向于先发生加氢反应,且不发生1-MN全加氢生成1-甲基十氢萘的反应。Wu T等[15]以1-MN为原料,M/Beta(M=NiW,NiMo,CoW,CoMo,W,Mo)为催化剂,在温度420 ℃和压力6 MPa条件下进行加氢裂化反应,结果表明,1-MN在该条件下不发生完全加氢再开环裂解的反应。
鞠雪艳等[16]在温度360 ℃、氢分压4.0 MPa和催化剂为NiMo/Al2O3条件下,通过实验和热力学计算研究1-MN加氢裂化反应。结果表明,1-MN中没有甲基取代的芳环优先发生加氢反应生成甲基四氢萘;而甲基四氢萘加氢生成甲基十氢萘反应的活化能远高于1-MN加氢生成甲基四氢萘。Sapre A V等[17]在温度325 ℃和压力7.0 MPa条件下,对萘在CoMo/γ-Al2O3上的加氢反应进行研究,结果表明,萘的第一个苯环的加氢反应速率快,而第二个苯环的加氢反应(即四氢萘的加氢反应)难以发生。Banerjee S等[18]研究表明,反应温度约400 ℃,稠环芳烃(如萘、1-MN等)第一个苯环的加氢反应比单环芳烃(如四氢萘、茚满和烷基苯等)的饱和加氢反应在热力学上更容易发生。在反应温度500 K(~227 ℃)时,单环芳烃(如苯)饱和加氢反应平衡常数比稠环芳烃(如萘、蒽)端环饱和加氢反应平衡常数大,但温度为700 K(~427 ℃)时,萘和蒽的端环加氢反应平衡常数和反应速率比苯饱和加氢反应平衡常数大。
因此,在加氢裂化反应条件下,双环芳烃的加氢是分步进行,一般优先发生无侧链苯环的饱和加氢反应得到单环芳烃,再发生与上节所述的四氢萘一致的反应。即四氢萘可直接异构、开环裂解,得到目标产物-轻质芳烃,也可能先发生饱和加氢生成十氢萘、再异构、开环裂解生成小分子烷烃。而双环芳烃是否发生完全加氢反应取决于反应条件和催化剂性质。
2.3 三环芳烃加氢裂化反应
目前研究者普遍认为三环芳烃(如蒽、菲及其同系物)的加氢反应是分步进行。Pinilla J L等[19]在反应温度(250~350) ℃和压力3.0 MPa条件下,采用Pd/Mesoporous Carbon beads(介孔碳微球)为催化剂研究蒽的加氢反应。部分蒽(a)发生中间苯环加氢生成对称二氢蒽(b);部分蒽发生端环加氢生成不对称四氢蒽(c)。二氢蒽和四氢蒽都可以进一步加氢生成非对称八氢蒽(e),而四氢蒽还可以加氢生成对称八氢蒽(d),两种八氢蒽皆可饱和加氢生成全氢蒽(f),但在实验条件下并未发现全氢蒽(f)的生成。Fan H等[20]以NiFe/HZSM-5为催化剂,在温度415 ℃和压力3.0 MPa下,研究蒽加氢裂化反应机理,提出3个转化途径:反应路径I,蒽(a)先发生中间苯环加氢生成对称二氢蒽(b),随后发生中间环的开环裂解生成两个烷基苯(g和h);反应路径Ⅱ,蒽先发生端环加氢生成不对称四氢蒽(c),四氢蒽再异构-开环裂解为烷基萘(i),烷基萘加氢生成烷基四氢萘(j),生成的烷基四氢萘又异构-开环裂解为烷基苯(h);反应路径Ⅲ,蒽(a)连续发生加氢反应生成对称八氢蒽(d),部分八氢蒽可异构化为对称八氢菲(k),生成的八氢菲通过氢转移反应生成二氢菲(l),蒽、二氢蒽和四氢蒽也可通过氢转移及异构化反应生成二氢菲,二氢菲最终裂解为乙基联苯(m)。实验条件下,蒽优先发生中间苯环加氢反应,反应产物以乙基联苯为主(收率为44%)。蒽加氢裂化反应路径如图3所示。
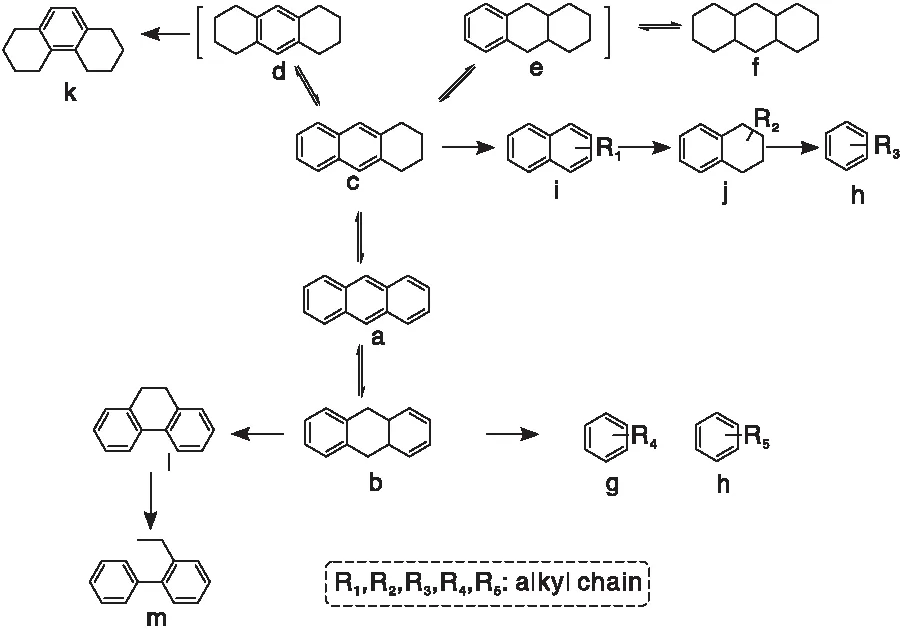
图3 蒽加氢裂化反应路径[20]
Chareonpanich M等[21]在温度(400~600) ℃和压力5 MPa条件下,以不负载金属的USY分子筛为催化剂,对稠环芳烃(蒽、菲、9,10-二氢菲)的加氢裂化进行研究。结果表明,菲首先发生中间苯环加氢反应,再发生端环的加氢、异构化、开环和裂解,并不发生中间环的加氢-开环-裂解生成烷基苯的反应。Korre S C等[22]在温度350 ℃和压力6.81 MPa下,以CoMo/Al2O3为催化剂,研究了菲的加氢反应速率,表明菲优先发生端环加氢生成双环芳烃,生成的二氢菲则优先在另一端环上发生加氢反应,并很快发生完全加氢反应生成全氢菲。Celis-Cornejo C M等[23]在反应温度380 ℃、压力6.9 MPa和空速1.3 h-1条件下,以NiMo/(Y + Al2O3)为催化剂,研究菲的三种加氢裂化反应路径:(Ⅰ)中间环加氢裂化;(Ⅱ) 菲的完全加氢反应再异构化、开环、裂解;(Ⅲ) 端环加氢裂化。在所使用实验条件下,菲优先发生路径Ⅲ的反应,即菲的一侧端环发生加氢裂解生成双环芳烃,或两个端环均发生加氢裂化生成单环芳烃。
三环芳烃在加氢裂化反应条件下或先发生一个端环加氢、异构、开环、裂解生成双环芳烃,之后发生的反应与上一节双环芳烃的反应路径一致。或先发生两个苯环的加氢反应,再异构、开环、裂解生成单环芳烃之后发生的反应与上一节双环芳烃的反应路径一致。在这种情况下,双环芳烃可以看作三环芳烃加氢裂化反应的中间产物[24]。三环芳烃也可能发生中间环加氢裂化反应生成两个单环芳烃分子,或发生三个苯环完全加氢再裂化生成小分子。
3 LCO加氢裂化催化剂
加氢裂化催化剂一般是由具有加氢活性的金属(或金属硫化物)和具有裂解活性的酸性载体组成的双功能催化剂。在加氢裂化反应条件下,LCO中的芳烃可能发生二类加氢裂化反应:一是芳烃部分加氢裂化反应;二是芳烃饱和加氢裂化反应。上述两类反应中仅第一类反应可得到轻质芳烃,生成的轻质芳烃也可能发生饱和加氢裂化生成小分子烷烃。因此,控制好芳烃的加氢深度(提高催化剂选择性加氢能力)会对提高轻质芳烃的收率起到至关重要的作用。催化剂性质(如加氢活性和酸性,是控制反应深度的关键因素[25])成为了科研工作者的研究重点。
3.1 加氢活性组分
活性金属一般采用贵金属(Pt、Pd、Ir等)或ⅥB族、Ⅷ族的过渡金属元素。其种类、负载量、分散度和不同金属的配比都会对其加氢活性产生影响[26-27]。
(1) 金属组分的种类及其用量
Ru和Rh是优良的加氢活性组分,但其加氢活性过高,产物中单环芳烃收率低;Pt和Pd等贵金属以及Mo和W等过渡金属的加氢活性适中,适用于多环芳烃的选择性加氢裂化制取BTX反应[28]。由于LCO中会存在一定量的含硫化合物,Pt和Pd等贵金属在催化其加氢反应时容易失活[29],因此,Mo和W等过渡金属是比较合适的加氢活性组分。Musser D M等[30]以萘为原料,在Raney Ni和Cu-Cr氧化物催化剂下分别进行加氢反应,结果表明,Raney Ni的催化反应活性虽然高于Cu-Cr氧化物,但产物四氢萘选择性低。Shin J等[31]认为加氢活性适中的CoMo/Beta和NiMo/Beta比NiW/Beta、Ni/Beta和NiSn/Beta更适于催化四氢萘加氢裂化制取BTX。
金属负载量与加氢反应速率直接相关。Wang W J等[32]研究了金属负载量与加氢活性的关系,发现适当提高金属负载量有利于提高加氢反应速率。但金属负载量过大时,会阻塞载体孔道,降低酸性中心的可接近性。
(2) 金属组分的分散度
一般认为,过渡金属硫化物堆垛的边、角位(brim位)金属原子具有较高的加氢活性,提高分散度即是提高金属的有效活性中心浓度。Ortega-Domínguez R A等[33]采用镍配合物制备Ni/SBA-15用于萘加氢反应,相较于传统前驱体制备的催化剂,萘 转化率由42.0%提高到96.3%(反应温度400 ℃,压力7.3 MPa,空时0.5 h),同时,十氢萘选择性也大幅提高。Tayeb K B等[34]采用Ni4SiW11O39水溶液(Keggin型缺位的杂多阴离子溶液)为前驱体制备NiW/Al2O3-SiO2双功能催化剂。对比传统催化剂,该催化剂金属分散度高,加氢活性高。Kim S H等[35]采用油溶性金属前驱体制备加氢裂化催化剂,形成的金属氧化物颗粒大小为(5~20) nm。Kim S H等[36]采用Mo(CO)6为前驱体,三辛基氧化膦为稳定剂,制备了高金属分散度的Mo/Al2O3-SiO2,用于减压渣油加氢裂化反应,显著提高了加氢活性。Morgado Prates A R等[37]采用四甲基溴化铵选择性去除Beta分子筛外表面聚集的Pt颗粒。相较于传统催化剂,甲苯加氢反应的转化率提高不明显,但活性中心效率提高了近10倍(TOF)。
(3) 金属组分的配比及助剂对加氢活性的影响
王永刚等[38]研究了NiW/γ-Al2O3中Ni/W原子比对低温煤焦油加氢反应的影响。当NiO的比例较低时,活性组分难以被还原,催化剂加氢活性差。而当Ni与W物质的量比为0.38时,加氢活性最高,同时具有较好的加氢脱氮、脱硫性能。Upare D P等[39]研究了Co/Mo原子比对石脑油裂解燃料油加氢裂化反应的影响。添加助剂Co时,载体表面形成CoMoO4活性相,金属的加氢活性显著提高。当Co/Mo原子比为0.5时,加氢活性最高,裂解燃料油的转化率达99.1%。但当Co/Mo原子比超过0.5时,金属的加氢活性反而下降,可能是由于CoMoO4活性相达到了饱和,同时载体的表面积下降。
适量助剂的加入可调变催化剂的酸量、酸强度及酸分布,同时也会对金属的分散度产生影响[40]。Liu X等[41]研究了Mg(0.5%~2%)和K(1%)的加入对Mo2C/HY催化萘加氢裂化反应的影响,Mg(以六水合硝酸镁为前驱体)的加入使得金属Mo分散度提高,但是对催化剂酸性影响不大。而K(以硝酸钾为前驱体)的加入改善了Mo分散度的同时,也使催化剂Brønsted酸量降低了30%,不利于开环反应的发生。关月明等[42]考察了助剂B对NiMo/Al2O3催化剂物化性质及加氢脱硫、氮活性的影响。加入少量的B,使催化剂Brønsted酸量增强,提高加氢脱硫、脱氮活性。但是加入过量的B,会使金属难以还原(金属与载体的相互作用太强),降低金属的利用率。
3.2 酸性组分-分子筛载体
在LCO加氢裂化反应过程中存在着异构化、开环和裂解等多种酸催化反应,不同的反应及反应物对催化剂酸性和孔道结构等的要求不尽相同[13],Y、Beta、ITQ-21和MCM-41等分子筛常被用作催化剂的酸性载体[43]。
(1) 分子筛的酸量、酸强度和酸类型
一般认为,四氢萘的异构-开环裂解反应及烷基苯的β断裂反应均发生在分子筛的B酸中心上[44-45]。Upare D P等[39]发现硅铝比低(SiO2与Al2O3物质的量比=25)的Beta制备的CoMo/Beta催化剂,含有大量的B酸,有利于促进开环和裂解反应,因此取得较高的BTX收率并表现出更好的稳定性。而Arribas M A等[46]认为,降低B酸量能够抑制1-MN加氢裂化中过度裂解反应的发生,从而提高轻质芳烃选择性。Ma H等[47]也发现在制备催化剂中加入一定量的K,催化剂的B酸量明显降低,四氢萘加氢裂化产物中单环芳烃收率明显提高,气体小分子产物明显减少。反应温度260 ℃时,Pt-K/USY(Pt质量分数0.5%,K质量分数2%)催化剂上C10烃类产物收率可保持在90%,烷基苯收率达35.6%。
Chareonpanich M等[48]发现在四氢化萘加氢裂化制取BTX反应中,以H-ZSM-5和H-Beta混合物为载体的催化剂,由于较强的酸性取得的BTX收率要高于以Al2O3和USY分子筛混合物为载体的催化剂所取得的BTX收率[31]。Kazakov M O等[49]认为适当降低催化剂B酸强度能够提高减压瓦斯油加氢裂化产物中间馏分油收率。Sato K等[9]使用不同的探针分子(蒽、菲、萘和四氢萘)对芳烃开环反应进行研究,结果表明,与二苯基甲烷断苯基链的裂化反应相比,四氢萘的开环过程需要的酸性较强。
(2) 分子筛的孔道结构
由于不同动力学尺寸的反应分子对活性中心的可接近性不同,因此选择合适的分子筛对提高产物选择性至关重要。研究表明,四氢萘和十氢萘分子可以进入ZSM-5分子筛的十元环孔道(0.55 nm)内,但其开环后的产物扩散受限,容易发生过度裂化[50]。菲可以进入Y分子筛的十二元环中进行加氢裂化反应,产物为苯、环烷烃及小分子烷烃等加氢裂化产物。但是菲不能进入丝光沸石的十元环和ZSM-5的十元环孔道,反应主要发生在分子筛的外表面,因而产物主要为二氢菲、四氢菲和八氢菲等加氢产物[51]。Tang J L等[52]在不同温度下分别采用ZSM-5和HY分子筛催化全氢菲(C14H24)开环裂解,发现在所选反应条件下(反应温度500 ℃,空速10 h-1),两种催化剂所得产物种类基本一致,为C1~C4烷烃、BTX等单环芳烃、重芳烃和环烷烃。HY催化活性更高,转化率为93%,而HZSM-5活性稍低,转化率为90%。但HZSM-5对芳烃选择性更高(68%),HY对芳烃选择性稍低(63%),且产生更多的重芳烃。这是由于HY相对HZSM-5具有更大的孔径及笼结构。Kim Y S等[53]分别以SiO2、ZSM-5、Beta和USY为载体制备加氢裂化催化剂,研究不同催化剂下萘的加氢裂化反应,结果显示,以Beta分子筛为载体的催化剂取得了最高的BTX收率。许多研究者在合成分子筛过程中添加介孔模板剂或采用后处理(酸处理、碱处理等)的方法制备介孔分子筛[54],以改善微孔分子筛对大分子的可接近性。Shin J等[31]以十六烷基三甲基溴化铵(CTAB)和四甲基氢氧化铵为介孔模板剂合成介孔Y分子筛。适当增大介孔体积,提高了反应物的转化率及BTX收率。安娟娟等[55]以CTAB为介孔模板剂合成介孔SAPO-5分子筛,制备了Ni/SAPO-5催化剂,用于催化萘选择性加氢反应,结果表明,随着分子筛介孔孔体积和孔径增加,萘转化率及十氢萘选择性均大幅提高。Imyen T等[56]采用碳纳米束作为介孔模板剂合成了介孔ZSM-5,并用于庚烷裂解反应。相比于普通ZSM-5,介孔ZSM-5的催化活性差别不大,但反应稳定性提高明显,这是由于介孔结构提高了催化剂容积炭量。Ren S等[57]先后采用NH4F和NaOH处理Y分子筛得到介孔Y分子筛(Meso-USY),用于萘加氢裂化反应。结果表明,NiMo/(Al2O3+Meso-USY)比NiMo/(Al2O3+USY)取得更高的烷基苯收率(提高18.5%)。
(3) 分子筛的颗粒粒径
Konno H等[58]研究了纳米级[(90~200) nm]和大颗粒(~2 300 nm)HZSM-5对甲基环己烷开环活性的影响。在反应温度450 ℃和压力1.0 MPa下,大颗粒HZSM-5催化活性较低,反应4.5 h,转化率从60%降到10%。而纳米级HZSM-5反应活性较高,且催化活性较稳定,反应45 h,转化率从90%降到83%。反应4 h后,大颗粒HZSM-5上积炭量为6.9%,而纳米级HZSM-5上为5.1%。
(4) 酸中心的化学环境(酸中心在分子筛骨架的分布)
Lee S U等[59]采用三种不同硅源合成Beta分子筛,并用于制备NiW/Beta催化剂。在催化甲基萘酚选择性开环反应中,以正硅酸乙酯为硅源合成的催化剂具有最好的催化活性,转化率为97%,单环芳烃收率达60%。不同硅源(正硅酸甲酯、正硅酸乙酯和正硅酸丁酯)的烷基链长短决定了Beta中Si元素的分布,进而决定了酸中心在骨架中的分布。
3.3 金属中心与酸中心的匹配
(1) 酸性中心与加氢活性中心的配比及相互作用
对于双功能催化剂,两种活性中心的配比及相互作用会影响其协同作用的效果,从而改变目标产物选择性[60]。Liu X等[61]通过调节金属负载量研究了两种活性中心的配比对萘加氢裂化反应的影响。当Mo2C负载量为20%时,HY和Mo2C之间的协同效果最好。此时,萘转化率达90%,加氢产物收率为58.5%,烷基苯收率达20.4%。当Mo2C负载量为7%时,萘转化率及加氢产物收率较低;当Mo2C负载量为27%时,烷基苯收率很低。D’Ippolito S A等[62]发现,金属的存在会影响HY分子筛的酸性质(酸量和酸强度)。在反应温度300 ℃条件下,铂与铱质量分数均为1.5%时,分子筛酸性适宜,两种活性中心协同效果最好,十氢萘的开环产物收率较高,达71.07%。Hengasawad T等[63]发现Pt物种在载体上分散均匀,且Y分子筛的B酸量较高时,两种活性中心达到了平衡,具有较高的催化生物柴油转化性能。
Qiu S等[64]通过向金属前驱体溶液中添加乙二醇调节金属-载体相互作用,研究其对萘加氢反应的影响。当乙二醇与Ni原子物质的量比为1时,金属与载体的相互作用适宜,形成的NiO分散度高(颗粒较小),有效活性位点多,使得萘可在55 ℃完全加氢生成十氢萘。Zhang B等[65]发现增强载体表面酸性有利于提高金属-载体相互作用,抑制金属的烧结,但也会降低金属加氢活性。Kim Y S等[66]在纳米级Beta(nano-Beta)和微米级Beta(micro-Beta)分子筛上负载Ni2P制备双功能催化剂,研究发现,Ni2P/nano-Beta催化剂上金属与分子筛相互作用更强,提高了催化剂的加氢活性及选择性,因而取得了比Ni2P/micro-Beta更高的1-MN转化率和BTX收率(42.3%)。Zhang S等[67]以CeO2纳米棒作为载体制备加氢催化剂,相较于SiO2或TiO2载体,该催化剂金属-载体相互作用更强,金属Pt的加氢活性弱,因而实现喹啉选择性加氢转化为1,2,3,4-四氢喹啉,选择性达80%。Van haandel L等[68]采用磷酸处理后的Al2O3作为加氢脱硫催化剂载体,结果表明,载体酸性减弱,降低了金属-载体相互作用,提高了金属还原度,增强了加氢脱硫活性。因此,增强载体酸性有利于提高金属分散度,增加有效活性中心浓度,但由于强金属-载体相互作用会导致金属氧化物难以还原,加氢活性受到抑制[69-70]。Wu T等[71]以钨配合物为金属前驱体制备了加氢裂化催化剂,在强酸性载体上实现了高加氢活性。
(2) 酸性中心与加氢活性中心的距离
金属中心与酸中心两种活性中心之间的匹配程度既受到两种活性中心数量是否匹配、相互作用是否适宜的影响,还受到距离上接近程度的影响[48,72]。Arribas M A等[28]研究了金属与载体的距离对四氢萘加氢裂化反应的影响(如图4)。研究表明,负载型催化剂的开环能力最强,单环芳烃的选择性随金属中心与酸中心距离缩短而提高。Samad J E等[73]将金属中心与酸中心距离分为原子尺度、纳米尺度、微米尺度和毫米尺度。通过设计具有不同活性中心距离的催化剂,研究正庚烷异构化反应。结果表明,金属中心与酸中心接近性介于纳米尺度及微米尺度的传统负载型催化剂反应活性高于物理混合型催化剂(毫米尺度)。Zecevic J等[74]采用Al2O3黏结剂调节Y分子筛与Pt的接近性,表明纳米尺度的可接近性有利于发挥两种活性中心的协同作用。Du H等[75]发现金属与酸中心距离越近,氢溢流效应越明显,催化活性越高。

图4 金属中心-酸中心可接近性模型
4 结语与展望
将劣质柴油-LCO中的芳烃转化为附加值高的轻质芳烃的工业实践,工艺流程较为复杂,轻质芳烃收率低。从稠环芳烃加氢裂化反应规律来看,控制芳烃的加氢反应深度是提高轻质芳烃收率的关键。目前研究主要集中在反应条件优化、加氢金属种类、负载量及分子筛载体的酸性质、孔道结构的调变,对催化剂选择性加氢能力关注较少,就金属的种类、负载量、分散度及分子筛性质对催化剂选择性加氢能力的影响尚没有清晰认识。同时对于LCO加氢裂化这个复杂反应过程,轻质芳烃收率取决于加氢活性中心和裂解活性中心的协同作用,目前尚不能定量确定两种活性中心在各步反应中的作用,因而不能得出确切的结论,也无法准确判断两种活性中心是否匹配。
