干鱼腥草药材及饮片的指纹图谱建立、化学计量学分析及含量测定
2022-04-27王静朱广飞梁思予张科卫陆兔林毛春芹
王静 朱广飞 梁思予 张科卫 陆兔林 毛春芹

中圖分类号 R917;R284.1 文献标志码 A 文章编号 1001-0408(2022)08-0923-07
DOI 10.6039/j.issn.1001-0408.2022.08.04
摘 要 目的 建立干鱼腥草药材及饮片的指纹图谱,并进行化学计量学分析,同时测定其中新绿原酸等5种黄酮类成分的含量。方法 采用高效液相色谱(HPLC)法,以槲皮苷为参照,绘制10批干鱼腥草药材及饮片的HPLC指纹图谱,采用《中药色谱指纹图谱相似度评价系统(2012版)》进行相似度评价,确定共有峰;采用SIMCA-P 14.1软件进行主成分分析(PCA)和偏最小二乘法-判别分析(PLS-DA),以变量重要性投影(VIP)值大于1为标准筛选影响干鱼腥草药材及饮片质量的差异性成分;采用同一HPLC法测定干鱼腥草药材及饮片中新绿原酸等5种成分的含量。结果 10批干鱼腥草药材及饮片均有20个共有峰,相似度均大于0.960;共指认了其中5个共有峰,分别为新绿原酸(1号峰)、绿原酸(3号峰)、隐绿原酸(4号峰)、芦丁(7号峰)、槲皮苷(11号峰)。PCA和PLS-DA结果均显示,干鱼腥草药材及饮片各自分为一类;VIP值大于1的共有峰依次为7号峰(芦丁)、20号峰、5号峰、13号峰、2号峰、18号峰、3号峰(绿原酸)、14号峰、17号峰和19号峰。新绿原酸、绿原酸、隐绿原酸、芦丁和槲皮苷检测质量浓度的线性范围分别为3.77~60.29 μg/mL(r=0.999 7)、1.40~22.42 μg/mL(r=0.999 5)、3.76~60.22 μg/mL(r=0.999 9)、2.19~35.06 μg/mL(r=0.999 9)、25.49~407.88 μg/mL(r=0.999 7);精密度、稳定性(24 h)、重复性试验的RSD均小于3%;干鱼腥草药材及饮片中上述5种成分的平均加样回收率分别为98.72%~101.12%、98.86%~100.63%(RSD均小于3%,n=9)。干鱼腥草药材中上述5种成分的平均含量分别为0.87、0.33、0.59、0.61、6.17 mg/g,饮片中分别为0.42、0.11、0.26、0.23、3.16 mg/g。结论 所建指纹图谱及含量测定方法稳定、可行,可用于干鱼腥草药材及饮片的质量控制;芦丁等成分可能是影响其药材及饮片质量的差异性成分;干鱼腥草药材经炮制后,新绿原酸等5种黄酮类成分的平均含量均有所降低。
关键词 干鱼腥草;药材;饮片;指纹图谱;含量测定;高效液相色谱法;化学计量学分析
Study on the fingerprint establishment, chemometrics analysis and content determination of dried Houttuynia cordata and its decoction pieces
WANG Jing,ZHU Guangfei,LIANG Siyu,ZHANG Kewei,LU Tulin,MAO Chunqin(School of Pharmacy, Nanjing University of Chinese Medicine, Nanjing 210023, China)
ABSTRACT OBJECTIVE To establish the fingerprints of dried Houttuynia cordata and its decoction pieces, conduct chemometrics analysis and determine the contents of 5 flavonoids such as neochlorogenic acid. METHODS High performance liquid chromatography (HPLC) method was adopted. Using quercitrin as reference, HPLC fingerprints of 10 batches of dried H. cordata and its decoction pieces were drawn. The similarity evaluation was conducted by Similarity Evaluation System of TCM Chromatographic Fingerprint (2012 edition), the common peaks were also confirmed. SIMCA-P 14.1 software was applied for principal component analysis (PCA) and partial least square-discriminant analysis (PLS-DA), and the variable importance in projection (VIP) value more than 1 was considered as a standard to screen the differential components affecting the quality of these two products; meanwhile, the contents of 5 components such as neochlorogenic acid in both products were determined by the same HPLC method. RESULTS There were 20 common peaks in 10 batches of dried H. cordata and 10 batches of its decoction pieces with the similarity values more than 0.960. A total of 5 common peaks were identified, which were neochlorogenic acid (peak 1), chlorogenic acid (peak 3), cryptochlorogenic acid (peak 4), rutin (peak 7) and quercitrin (peak 11). The results of PCA and PLS-DA showed that dried H. cordata could be distinguished from its decoction pieces obviously; the common peaks with VIP value greater than 1 were as follows: peak 7 (rutin), peak 20, peak 5, peak 13, peak 2, peak 18, peak 3 (chlorogenic acid), peak 14, peak 17 and peak 19. The linear range of neochlorogenic acid, chlorogenic acid, cryptochlorogenic acid, rutin and quercitrin were 3.77-60.29 μg/mL (r=0.999 7), 1.40-22.42 μg/mL (r=0.999 5), 3.76-60.22 μg/mL (r=0.999 9), 2.19-35.06 μg/mL (r=0.999 9) and 25.49-407.88 μg/mL (r=0.999 7), respec- tively. RSDs of precision, stability (24 h) and reproducibility tests were all lower than 3%. The average recoveries of the above components in these two products were 98.72%-101.12% and 98.86%-100.63% with RSDs less than 3% (n=9). In dried H. cordata, the average contents of 5 components were 0.87, 0.33, 0.59, 0.61 and 6.17 mg/g, while the average contents were 0.42, 0.11, 0.26, 0.23 and 3.16 mg/g in its decoction pieces, respectively. CONCLUSIONS HPLC fingerprint and the method of content determination are stable and feasible, which could be used for the quality control of dried H. cordata and its decoction pieces. Besides, rutin and other components may be the differential components which could affect the quality of these two products; the average contents of the 5 flavonoids such as neochlorogenic acid in dried H. cordata all decrease after processing.
KEYWORDS dried Houttuynia cordata; medicinal material; decoction pieces; fingerprint; content determination; high performance liquid chromatography; chemometrics analysis
鱼腥草为三白草科植物蕺菜Houttuynia cordata Thunb.的新鲜全草或干燥地上部分,其味辛,性微寒,归肺经,具有清热解毒、消痈排脓、利尿通淋的功效[1]。该药主要含有挥发油类、黄酮类等成分,具有抗炎、抑菌、抗病毒、抗肿瘤等作用,常与其他中药配伍用于呼吸道感染、扁桃体炎、流行性腮腺炎、角膜炎、病毒性肠炎、鼻咽癌等的治疗[2-4]。目前,含鱼腥草的成方制剂和单味制剂使用广泛,如被2020年版《中国药典》(一部)收录的连花清瘟胶囊(颗粒)、鱼腥草滴眼液、复方鱼腥草片(合剂)、急支糖浆等。鱼腥草的化学成分复杂,2020年版《中国药典》(一部)“鱼腥草”项下含鲜鱼腥草和干鱼腥草,但质量控制均仅以外观性状、显微、薄层色谱、理化鉴别为主[1],缺乏全面反映其质量的客观指标及含量测定项。笔者查阅文献发现,含鱼腥草的眼用制剂主要以其挥发油类成分(甲基正壬酮、鱼腥草素)为质量控制指标[1,5],而口服制剂则多以黄酮类成分(新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷等)为质量控制指标[6-8],且上述黄酮类成分亦为鱼腥草中含量较高的活性成分[9-10]。考虑在实际大工业生产中,所用原料基本均为干鱼腥草药材及饮片,因此本研究拟对干鱼腥草药材及饮片中的黄酮类成分(新绿原酸、绿原酸、隐绿原酸、芦丁和槲皮苷)进行分析。
中药指纹图谱是控制中药及其制剂质量的有效手段,可较全面地反映中药及其制剂中化学成分的种类与含量,可对其质量进行全面表征并实现对多种药效成分的综合评价[11]。化学计量学是筛选中药质量标志物的重要数学方法,具有较好的预测精度和广泛的适用性,可分为非监督模式识别方法和监督模式识别方法,前者主要包括主成分分析(principal componet analysis,PCA)、聚类分析等,后者主要包括偏最小二乘法-判别分析(partial least square-discrimination analysis,PLS-DA)、人工神经网络等[12]。指纹图谱结合化学计量学分析,能有助于研究者对指纹图谱的有效信息进行提取和处理,现已被广泛应用于中药材及饮片的鉴别和质量评价[11-12]。基于此,本研究拟建立干鱼腥草药材及饮片的高效液相色谱(high performance liquid chromatography,HPLC)指纹图谱,并结合化学计量学方法分析其药材与饮片的差异;与此同时,拟采用同一HPLC法测定干鱼腥草药材及饮片中新绿原酸、绿原酸、隐绿原酸、芦丁和槲皮苷5种黄酮类成分的含量,旨在为全面控制其整体质量、进一步开发相关制剂提供依据。
1 材料
1.1 主要仪器
本研究所用主要仪器有LC-20AD型HPLC仪及配备的DGC-20A型在线脱气系统、SIL-20A型自动进样系统、CTO-20AC型柱温箱、SPD-M20A型二极管阵列检测器(日本Shimadzu公司),MS-105D型十万分之一电子分析天平(瑞士Mettler Toledo公司),FA1104N型电子天平(上海精密科学仪器有限公司)等。
1.2 主要药品与试剂
新绿原酸对照品(批号DSTDX001503)、隐绿原酸对照品(批号DSTDY003502)均购自成都乐美天医药科技有限公司,绿原酸对照品(批号110753-202018)购自中国食品药品检定研究院,芦丁对照品(批号Y06J8S37439)、槲皮苷对照品(批号18082102)均购自上海源叶生物科技有限公司,纯度均大于98%;乙腈、乙酸为色谱纯,其余试剂均为分析纯,水为纯净水。
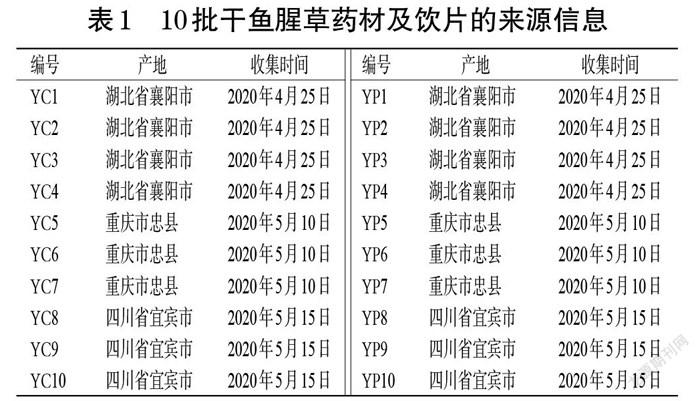
10批干鱼腥草药材(编号YC1~YC10)及10批饮片(编号YP1~YP10)均购自湖北省襄阳市、重庆市忠县、四川省宜宾市的中药材市场,经南京中医药大学药学院陈建伟教授鉴定为三白草科植物蕺菜H. cordata Thunb.的干燥地上部分。10批干鱼腥草药材及饮片的来源信息见表1。
2 方法与结果
2.1 干魚腥草药材及饮片HPLC指纹图谱的建立
2.1.1 色谱条件 以Merck Purospher Star LP RP-18 Endcapped(250 mm×4.6 mm,5 μm)为色谱柱,以乙腈(A)-0.05%乙酸溶液(B)为流动相进行梯度洗脱(0~26 min,8%A;26~31 min,8%A→14%A;31~61 min,14%A→24%A,61~86 min,24%A→39%A;86~104 min,39%A→74%A;104~108 min,74%A→95%A);流速为1.0 mL/min;检测波长为320 nm;柱温为25 ℃;进样量为10 μL。
2.1.2 混合对照品溶液的制备 精密称取新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷对照品适量,加甲醇溶解并稀释,制成上述5种成分质量浓度分别为120.576、44.832、120.448、70.128、815.760 μg/mL的混合对照品溶液。
2.1.3 供试品溶液的制备 取样品粉末(过三号筛,下同)约1.0 g,精密称定,置于圆底烧瓶中,精密加入70%乙醇30 mL,称定质量,加热回流30 min,放冷,再次称定质量,用70%乙醇补足减失的质量,摇匀,滤过,取续滤液,经0.45 μm微孔滤膜滤过,即得。
2.1.4 精密度试验 取干鱼腥草药材(编号YC1)、干鱼腥草饮片(编号YP1)样品粉末,按“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件连续进样测定6次,记录色谱图。以槲皮苷为参照,计算各共有峰的相对保留时间和相对峰面积。结果显示,干鱼腥草药材中各共有峰相对保留时间的RSD为0.22%~0.97%(n=6),相对峰面积的RSD为0.98%~1.64%(n=6);干鱼腥草饮片中各共有峰相对保留时间的RSD为0.23%~1.14%(n=6)、相对峰面积的RSD为1.09%~1.84%(n=6),表明方法精密度良好。
2.1.5 稳定性试验 取干鱼腥草药材(编号YC1)、干鱼腥草饮片(编号YP1)样品粉末,按“2.1.3”项下方法制备供试品溶液,分别于室温下放置0、2、4、8、12、24 h时按“2.1.1”项下色谱条件进样测定,记录色谱图。以槲皮苷为参照,计算各共有峰的相对保留时间和相对峰面积。结果显示,干鱼腥草药材中各共有峰相对保留时间的RSD为0.39%~1.21%(n=6),相对峰面积的RSD为1.62%~2.39%(n=6);干鱼腥草饮片中各共有峰相对保留时间的RSD为0.56%~1.20%(n=6),相对峰面积的RSD为1.78%~2.54%(n=6),表明供试品溶液于室温下放置24 h内稳定性良好。
2.1.6 重复性试验 取干鱼腥草药材(编号YC1)、干鱼腥草饮片(编号YP1)样品粉末,各6份,按“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样测定,记录色谱图。以槲皮苷为参照,计算各共有峰的相对保留时间和相对峰面积。结果显示,干鱼腥草药材中各共有峰相对保留时间的RSD为1.07%~1.82%(n=6),相对峰面积的RSD为1.01%~2.10%(n=6);干鱼腥草饮片中各共有峰相对保留时间的RSD为0.99%~1.83%(n=6),相对峰面积的RSD为1.02%~2.23%(n=6),表明方法重复性良好。
2.1.7 指纹图谱的建立 分别取10批干鱼腥草药材及饮片样品粉末,按“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样测定,记录色谱图。将上述色谱图导入《中药色谱指纹图谱相似度评价系统(2012版)》,以任意样品的色谱图为参照图谱,采用中位数法,设置时间窗宽度为0.1 min,进行多点校正和色谱峰匹配,经全峰匹配后得到干鱼腥草药材及饮片的叠加指纹图谱和对照指纹图谱(R)。结果显示,10批干鱼腥草药材及饮片均有20个共有峰,详见图1、图2。
2.1.8 共有峰的指认 取“2.1.2”项下混合对照品溶液,按“2.1.1”项下色谱条件进样测定,记录色谱图。将所得色谱图(图3A)与“2.1.7”项下干鱼腥草药材及饮片的色谱图进行比对,共指认了5个共有峰,分别为新绿原酸(1号峰)、绿原酸(3号峰)、隐绿原酸(4号峰)、芦丁(7号峰)、槲皮苷(11号峰)。因槲皮苷的峰面积较大且周围基线平稳,干扰峰较少,故选择槲皮苷为参照。此外,与干鱼腥草药材相比,其饮片样品色谱中的5号峰和14号峰的峰面积均显著增加,可用于区分干鱼腥草药材和饮片。
2.1.9 相似度评价 采用《中药色谱指纹图谱相似度评价系统(2012版)》对干鱼腥草药材及饮片指纹图谱的相似度进行评价。结果显示,与对照指纹图谱比较,干鱼腥草药材及饮片指纹图谱的相似度均大于0.960,表明各样品化学成分的种类一致性良好,详见表2。
2.2 化学计量学分析
2.2.1 PCA 为探讨干鱼腥草药材及饮片的成分差异,本研究以10批干鱼腥草药材及10批饮片共有峰的相对峰面积为变量,采用SIMCA-P 14.1软件进行PCA,结果见图4。由图4可知,干鱼腥草药材及饮片的主成分空间分布有各自特定的区域,沿主成分1轴左右分开,其中干鱼腥草药材分布于得分图的右侧,其饮片分布于得分图的左侧,表明两者化学成分存在差异。
2.2.2 PLS-DA 为更好地观察组间差异,本研究以10批干鱼腥草药材及饮片共有峰的相对峰面积为变量,采用SIMCA-P 14.1软件进行PLS-DA,结果见图5。由图5可知,模型的累积解释度(R 2X、R 2Y)分别为0.582、0.843,累积预测度(Q 2)为0.669,均大于0.5,表明所建数学模型可靠[13]。干鱼腥草药材及饮片各自分为一类,沿主成分1轴明显分开,其中干鱼腥草药材分布于得分图的左侧,其饮片分布于得分图的右侧,与上述PCA结果基本一致。
为明确影响干鱼腥草药材及饮片质量的差异性成分,本研究以10批干鱼腥草药材及饮片共有峰的相对峰面积为变量,采用SIMCA-P 14.1软件对各共有峰对应成分的变量重要性投影(variable importance in projection,VIP)值進行分析,以VIP值>1为标准筛选出贡献较大的差异性成分[12],结果见图6。由图6可知,VIP值>1的共有峰依次为7号峰(芦丁)、20号峰、5号峰、13号峰、2号峰、18号峰、3号峰(绿原酸)、14号峰、17号峰和19号峰,这些共有峰对应的成分可能是影响干鱼腥草药材及饮片质量的差异性成分。
2.3 含量测定
2.3.1 色谱条件 同“2.1.1”项。
2.3.2 混合对照品溶液的制备 同“2.1.2”项。
2.3.3 供试品溶液的制备 同“2.1.3”项。
2.3.4 空白溶液的制备 不加样品,按“2.3.3”项下“精密加入70%乙醇30 mL……即得”操作,即得空白溶液。
2.3.5 系统适用性试验 取上述混合对照品溶液、供试品溶液、空白溶液适量,按“2.3.1”项下色谱条件进样测定,记录色谱图(图3)。由图3可知,各成分分离度均大于1.5,理论板数均不低于3 000,空白溶液对各成分的测定无干扰。
2.3.6 线性关系考察 取“2.3.2”项下混合对照品溶液适量,用甲醇稀释,制成新绿原酸质量浓度分别为3.77、7.54、15.07、30.14、60.29 μg/mL,绿原酸分别为1.40、2.80、5.60、11.21、22.42 μg/mL,隐绿原酸分别为3.76、7.53、15.06、30.11、60.22 μg/mL,芦丁分别为2.19、4.38、8.77、17.53、35.06 μg/mL,槲皮苷分别为25.49、50.99、101.97、203.94、407.88 μg/mL的系列工作溶液,按“2.3.1”项下色谱条件进样测定,记录色谱图。以各待测成分质量浓度(X)为横坐标、峰面积(Y)为纵坐标进行线性回归,结果见表3。
2.3.7 精密度试验 取“2.3.6”项下某系列工作溶液(新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷质量浓度分别为15.07、5.60、15.06、8.77、101.97 μg/mL)适量,按“2.3.1”项下色谱条件连续进样测定6次,记录峰面积。结果显示,新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷峰面积的RSD分别为0.55%、1.01%、1.11%、0.94%、0.13%(n=6),表明仪器精密度良好。
2.3.8 稳定性试验 取“2.3.3”项下干鱼腥草药材(编号YC1)、干鱼腥草饮片(编号YP1)供试品溶液适量,分别于室温下放置0、2、4、8、12、24 h时按“2.3.1”项下色谱条件进样测定,记录峰面积。结果显示,干鱼腥草药材中新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷峰面积的RSD分别为1.06%、1.28%、1.69%、1.86%、0.09%(n=6),干鱼腥草饮片中新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷峰面积的RSD分别为1.14%、1.33%、1.75%、2.30%、0.12%(n=6),表明上述供试品溶液于室温下放置24 h内稳定性良好。
2.3.9 重复性试验 取干鱼腥草药材(编号YC1)、干鱼腥草饮片(编号YP1)样品粉末,各6份,按“2.3.3”项下方法制备供试品溶液,再按“2.3.1”项下色谱条件进样测定,记录峰面积并按标准曲线法计算样品中各待测成分的含量。结果显示,干鱼腥草药材中新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷含量的RSD分别为1.98%、1.68%、1.73%、2.67%、0.20%(n=6),干鱼腥草饮片中新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷含量的RSD分别为1.72%、1.56%、1.93%、2.41%、0.31%(n=6),表明方法重复性良好。
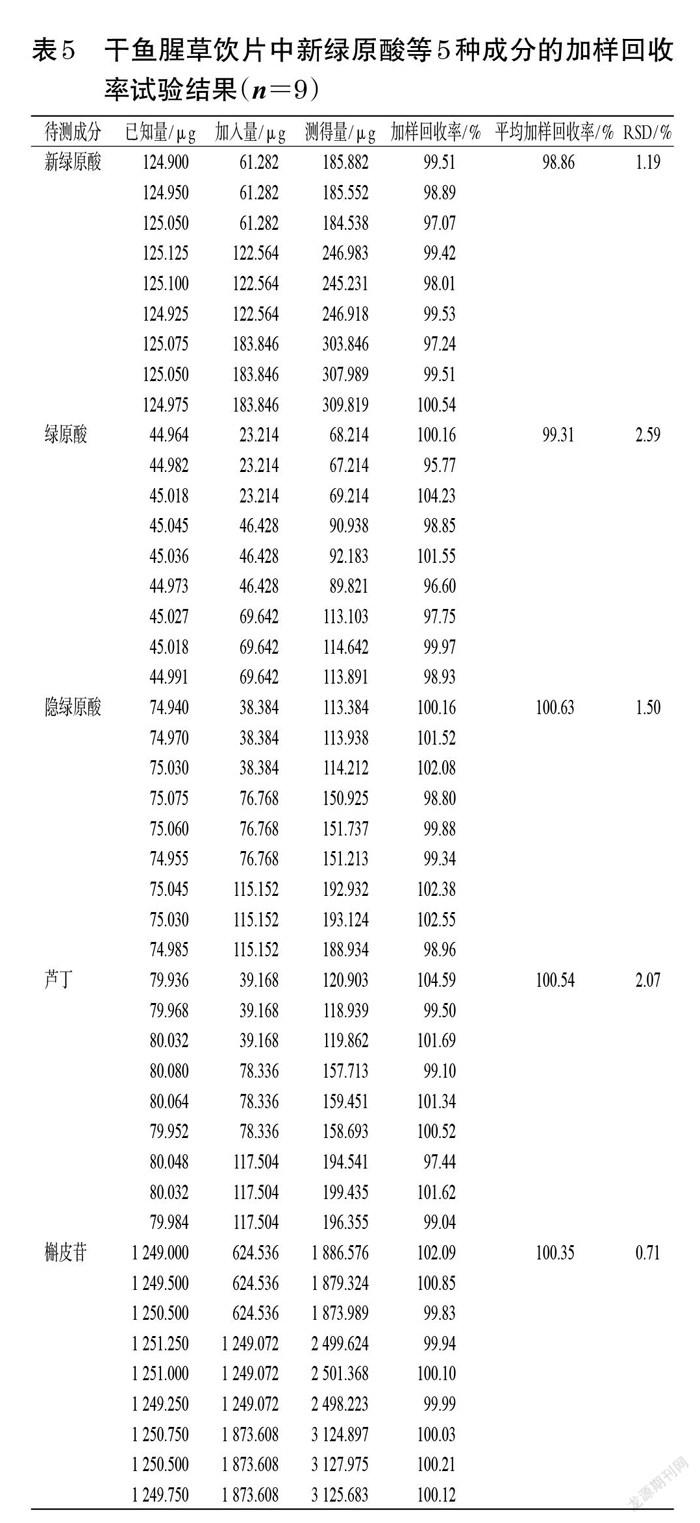
2.3.10 加样回收率试验 精密称取已知含量的干鱼腥草药材(编号YC1)样品粉末0.5 g,共9份,分别加入混合对照品溶液(精密称取新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷对照品适量,加甲醇溶解并稀释,制成各成分质量浓度分别为490.256、278.568、364.648、293.760、3 065.904 μg/mL的混合对照品溶液)0.5、1.0、1.5 mL,按“2.3.3”项下方法制备供试品溶液,再按“2.3.1”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表4。精密称取已知含量的干鱼腥草饮片(编号YP1)样品粉末0.5 g,共9份,分别加入混合对照品溶液(精密称取新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷对照品适量,加甲醇溶解并稀释,制成各成分质量浓度分别为122.564、46.428、76.768、78.336、1 249.072 μg/mL的混合对照品溶液)0.5、1.0、1.5 mL,按“2.3.3”项下方法制备供试品溶液,再按“2.3.1”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表5。
2.3.11 样品含量测定 取10批干鱼腥草药材及饮片样品粉末,按“2.3.3”项下方法制备供试品溶液,再按“2.3.1”项下色谱条件进样测定,记录峰面积并按标准曲线法计算样品含量。每样品平行测定3次,结果见表6。由表6可知,将干鱼腥草药材炮制为饮片后,新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷的平均含量均不同程度地降低,其中芦丁的降幅最大,约为62.3%。
3 討论
本课题组前期分别对供试品溶液的提取溶剂、提取方式、料液比和加热回流时间进行了考察,结果显示,当以70%乙醇为提取溶剂、料液比为1 ∶ 30(g/mL)、加热回流时间为30 min时,提取效率最高,所得色谱图的基线较平稳、各色谱峰分离度较好且杂质干扰较小。综合考虑,最终确定供试品溶液的制备方法如下:取样品粉末约1.0 g,加入70%乙醇30 mL,加热回流30 min。本课题组前期采用二极管阵列检测器在190~800 nm波长范围内进行扫描,结果显示,待测样品在320 nm波长下所得色谱信息较全面,色谱峰峰形整体良好,故选择检测波长为320 nm。此外,本课题组前期还考察了乙腈-水、乙腈-0.05%磷酸溶液、乙腈-0.05%乙酸溶液3个流动相体系的分离效果,结果显示,当以乙腈-0.05%乙酸溶液为流动相时,各色谱峰分离效果较好且杂质干扰较小。
目前,关于鱼腥草的研究大多为鱼腥草药材,且仅限于指纹图谱研究[14-16]或含量测定[17-21],而两者结合的研究较少。虽然,张婷婷等[22]、卢红梅等[23]将定性与定量分析相结合,但只研究了其中1~3种黄酮类成分,较为局限,不能全面反映鱼腥草的质量。何兵等[10]虽然在建立鱼腥草药材指纹图谱的基础上,采用一测多评法测定了其中多个成分的含量,但该方法对实验条件的要求较高。同时,笔者通过查阅文献发现,尚少有关于鱼腥草饮片的研究,考虑到鲜鱼腥草在采集和储存方面存在一定困难,且制剂生产所用原料多为干鱼腥草药材及饮片,因此本研究以干鱼腥草药材及饮片为对象,建立了两者的HPLC指纹图谱。结果显示,10批干鱼腥草药材及饮片均有20个共有峰,与对照指纹图谱的相似度均大于0.960,表明干鱼腥草药材经炮制后,其化学成分的种类未发生明显改变。笔者在分析各共有峰的峰面积时发现,干鱼腥草药材经炮制后,其5号峰和14号峰的峰面积明显增加,推测其原因可能与水处理和加热(干燥)过程有关。因此,笔者认为,上述两色谱峰的峰面积可用于区分干鱼腥草药材和饮片。PCA和PLS-DA结果均显示,干鱼腥草药材和饮片各自分为一类,7号峰(芦丁)、20号峰、5号峰、13号峰、2号峰、18号峰、3号峰(绿原酸)、14号峰、17号峰和19号峰对应的成分可能是影响干鱼腥草药材及饮片质量的差异性成分。
含量测定结果显示,干鱼腥草药材中新绿原酸等5种成分的含量分别为0.29~1.68、0.15~0.67、0.23~1.44、0.43~0.95、3.82~8.74 mg/g,其平均含量分别为0.87、0.33、0.59、0.61、6.17 mg/g;干鱼腥草饮片中上述5种成分的含量分别为0.19~0.60、0.05~0.20、0.15~0.38、0.12~0.38、2.00~4.87 mg/g,其平均含量分别为0.42、0.11、0.26、0.23、3.16 mg/g,表明干鱼腥草药材经炮制后,新绿原酸、绿原酸、隐绿原酸、芦丁、槲皮苷的平均含量均有不同程度的降低,其原因可能为炮制过程中的水洗和干燥步骤使上述成分有所损失[24-25]。
综上所述,本研究所建指纹图谱及含量测定方法稳定、可行,可用于干鱼腥草药材及饮片的质量控制;芦丁等成分可能是影响其药材及饮片质量的差异性成分;干鱼腥草药材经炮制后,新绿原酸等5种黄酮类成分的平均含量均有所降低。
参考文献
[ 1 ] 国家药典委员会.中华人民共和国药典:一部[S]. 2020年版.北京:中国医药科技出版社,2020:234-235,1191-1192.
[ 2 ] 麦明朗,余林中,刘俊珊.“中药抗生素”鱼腥草抗炎作用研究及临床应用进展[J].中药药理与临床,2018,34(5):172-176.
[ 3 ] 陆晓珊,林也,唐琳,等.鱼腥草的化学成分与安全性研究进展[J].中华中医药学刊,2021,39(3):144-147.
[ 4 ] 蔡红蝶,刘佳楠,陈少军,等.鱼腥草化学成分、生物活性及临床应用研究进展[J].中成药,2019,41(11):2719- 2728.
[ 5 ] 李明娟,朱琪,刘丰高,等. HPLC测定鱼腥草滴眼液中的鱼腥草素[J].华西药学杂志,2014,29(6):731-732.
[ 6 ] 陈育鹏,赵倩,王贵金,等.连花清瘟胶囊超高效液相指纹图谱研究[J].中国中医药信息杂志,2017,24(12):77-80.
[ 7 ] 王发英,吴查青,陈张金,等.复方鱼腥草合剂中3个有效成分及防腐剂的含量测定[J].中国现代应用药学,2018,35(3):399-403.
[ 8 ] 刘彬果.复方鱼腥草制剂的质量控制研究[D].上海:第二军医大学,2004.
[ 9 ] 郑亚娟,彭秋实,马义虔,等.鱼腥草化学成分的研究进展[J].广东化工,2017,44(17):85-86.
[10] 何兵,刘艳,田吉,等.指纹图谱结合一测多评模式在中药鱼腥草质量评价中的应用研究[J].中国中药杂志,2013,38(16):2682-2689.
[11] 明凯利,凌骅,向阳,等.健脾生血颗粒HPLC指纹图谱研究[J].中国药师,2021,24(11):2111-2114.
[12] 张浩杰,李焕茹,冯志毅,等.女贞子与酒女贞子HPLC指纹图谱对比研究[J].中国中药杂志,2020,45(16):3871- 3876.
[13] 刘宏,宋奇,王添敏,等.胡桃楸枝和根中5个成分含量测定方法的建立及其含量差异分析[J].中國药房,2021,32(8):933-939.
[14] 应军,苏流坤.鱼腥草HPLC指纹图谱的研究[J].中药材,2004,27(10):737-738.
[15] 李晓蒙,徐位良,何新荣.鱼腥草药材中黄酮苷类成分的HPLC指纹图谱研究[J].广东药学院学报,2005,21(2):140-142,152.
[16] 李转花,胡思玉,余启波,等.鲜鱼腥草高效液相色谱指纹图谱研究[J].甘肃中医药大学学报,2016,33(6):38-42.
[17] 李永伟,何兵,周德兵,等.高效液相色谱法同时测定鱼腥草中绿原酸和金丝桃苷含量[J].中国药业,2015,24(7):49-51.
[18] 印晓红,金汉台,谭林威,等.不同产地鱼腥草中槲皮苷含量测定[J].中国中医药信息杂志,2016,23(12):78-80.
[19] 张思荻,赖月月,杨超,等.基于金丝桃苷和槲皮苷的鱼腥草含量测定及质量分析[J].中国现代中药,2018,20(5):556-560,569.
[20] 信文远,张侠,李晓艳,等. HPLC法测定鱼腥草中槲皮苷的含量[J].中医药导报,2020,26(15):56-58.
[21] 廖卫波,王亚敏,戴迪,等. HPLC法同时测定鱼腥草中4种黄酮苷的含量[J].实用中西医结合临床,2020,20(11):157-159.
[22] 张婷婷,吴毅,杭太俊.鱼腥草黄酮类成分HPLC指纹对照法研究[J].中药材,2009,32(5):687-690.
[23] 卢红梅,彭丽华,郭方遒,等.鱼腥草中黄酮类成分的高效液相色谱指纹图谱分析[J].色谱,2010,28(10):965-970.
[24] 孙阳,黄和,胡燚.绿原酸提取纯化方法的研究进展[J]. 化学试剂,2017,39(3):257-262.
[25] 刘永玲,马风伟,赵治兵,等.野地瓜茎中3种绿原酸提取工艺的优化及抗氧化活性研究[J].食品研究与开发,2021,42(18):100-106.
(收稿日期:2021-09-29 修回日期:2022-01-20)
(编辑:陈 宏)
