Fe1-xO基氨合成催化剂助催化剂的优选
2022-04-26孙珍珍刘化章叶攀韩文锋
孙珍珍,刘化章,叶攀,韩文锋
(浙江工业大学工业催化研究所,浙江 杭州 310014)
基于合成氨的化肥是确保国家粮食安全的物质基础,而粮食的战略地位又决定了合成氨工业的重要性。合成氨生产的原料和燃料都是能源,是高耗能工业之一,节能潜力大。而催化剂在合成氨工业的节能减排、转型升级等方面起着关键作用。FeO 基氨合成催化剂是我国独创的新一代熔铁催化剂,因具有活性高、稳定性好、价格低廉、制备方法简单等优点被工业上广泛应用。因此进一步提高FeO基氨合成催化剂活性有重大意义。
由于FeO 基氨合成催化剂采用熔融法制备,活性组分和制备方法已经被固定,且不需要载体,因此影响催化剂性能的关键因素是助催化剂,助催化剂对FeO基催化剂活性和还原性能、耐热性能等方面有显著影响。也就是说,想要进一步提高FeO基催化剂的活性,助催化剂的研究是关键。但是助催化剂种类繁多,物化性质不同,对催化剂的性能影响不同,这是目前研究FeO基氨合成催化剂面临的重要挑战,因此助催化剂的筛选和优化必不可少。本文作者课题组前期工作中,已经对40 多种助催化剂进行了系统研究。本文在前期研究的基础上,研究了MgO、VO、ZrO、WO四种助催化剂及其交互作用,分别得到四、五、六和七助催化剂的FeO基催化剂的最优配方,并探究其高活性的原因。
1 实验部分
1.1 催化剂的制备
采用高温熔融法制备催化剂。首先根据催化剂配方,准确称取磁铁矿粉、还原铁粉和各助催化剂组分的质量,并在研钵中研磨混合均匀;接着将混合物料置于电阻炉中,用2根铁丝点火引燃,物料全部熔融后,将高温熔浆迅速流入水夹套式的冷却槽中冷却至室温,再经过破碎、筛分至所需要颗粒大小,装入广口瓶中作好标记。
1.2 催化剂的表征
程序升温还原(H-TPR)在GC1690 气相色谱中进行。催化剂在He 气氛条件下,从室温以5℃/min的升温速率升到200℃预处理1h,待冷却到室温后,在30mL/min的H(5%)/Ar(95%)混合气流中,以10℃/min 的升温速率升到850℃进行程序升温还原。
程序升温脱附(N-TPD)在上述装置中进行。催化剂在450℃下用纯H还原5h,然后降至350℃,通入N反应2h后,再用He气吹扫2h,之后再通入N吸附2h至饱和,缓慢降温至40℃,再通He气吹2h 去除催化剂表面物理吸附的N,在40~750℃范围内,以10℃/min 程序升温脱附,He 气流量为25mL/min。
催化剂物相,采用瑞士Thermo ARL X’TRA型X 射线衍射仪测定。CuK光(=1.54056Å,1Å=0.1nm),电压45kV,电流40mA,扫描速率2°/min。
N低温物理吸附实验在Micromeritics 公司的ASAP2010 型吸附仪上进行。还原后的催化剂先在200℃真空条件下脱气8h,然后在液氮温度下进行N吸附脱附实验。
SEM-EDS 采用HitachiS-4700Ⅱ型扫描电子显微镜对催化剂进行扫描,工作电压为15kV,并用Thermo Noran Vantage ESTX 射线能谱仪测定催化剂表面各元素的含量及其分布。
1.3 催化剂活性评价
活性评价按HG/T 3545—2014《氨合成催化剂试验方法》执行,在双同体八管反应器高压装置中进行,在完全相同的实验条件下可以同时测定8只样品。反应器内径为14mm,装填粒度为1.0~1.4mm 的催化剂2mL。在活性检测前,催化剂在400℃、425℃、450℃、475℃、500℃下程序升温还原31h,H、N摩尔比为3∶1,压力5MPa、空速30000h。还原结束后降温至活性测定温度(400℃、425℃、450℃),在压力15MPa、空速30000h条件下测定初活性;之后催化剂在500℃下耐热24h,测定耐热后的活性。反应器出口氨浓度用酸碱中和法测定。
2 结果与讨论
2.1 催化剂活性和耐热性
本实验中,固定AlO、KO、CaO 三个基础助催化剂,针对MgO、VO、ZrO、WO四种助催化剂(所有助剂的添加量都为前期优化出的最佳量),采用简单对比法,设计单变量(图1)、双变量(图2)、三变量和四变量(图3)实验方案,逐步考察各变量对催化剂性能的影响,直至优选出最佳催化剂。例如图1 中S-1(无)表示除了AlO、KO、CaO 助催化剂外,不含有其他组分,而S-2(MgO)则表示除了AlO、KO、CaO 助催化剂外,还含有MgO,余类推。MgO、VO、ZrO、WO是前期研究得到的较优良的助催化剂,催化剂活性评价结果如图1~图3和表1所示。

表1 催化剂耐热前后反应器出口氨浓度(评价条件:15MPa、30000h-1;耐热条件:500℃、24h) 单位:%
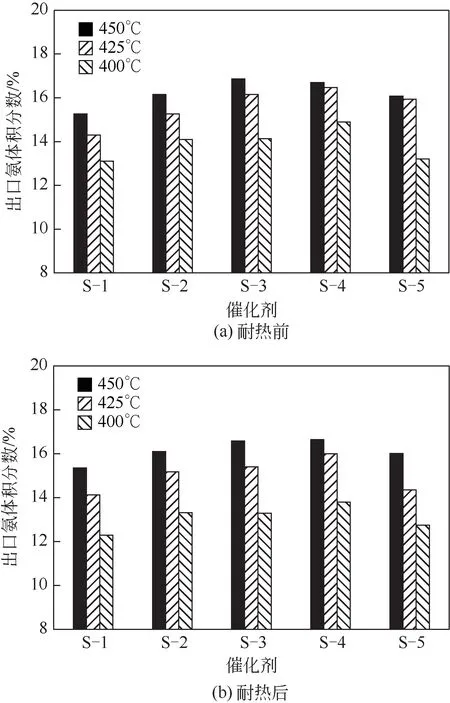
图1 各催化剂耐热前和耐热后的氨合成活性

图2 各催化剂耐热前和耐热后的氨合成活性

图3 各催化剂耐热前和耐热后的氨合成活性
图1 是(S-1)~(S-5)五个样品耐热前后的活性评价结果。由图1 可知,(S-2)~(S-5)四个样品耐热前后活性都高于S-1催化剂,表明MgO、VO、ZrO、WO助催化剂都可以不同程度地提高催化剂的活性,且450℃和425℃的活性呈现火山形曲线,其中耐热前的活性S-3 略优于S-4,但耐热后的活性则S-4优于S-3,说明VO有利于提高催化剂的活性,而ZrO则有利于提高催化剂的耐热性。
图2是(S-6)~(S-11)六个样品耐热前后的活性评价结果。由图2可知,由单变量(图1)改为双变量后,各催化剂活性都提高了一个档次(425℃为17%),其中,耐热前的活性除S-8较低外,其余难分伯仲;而耐热后含有VO、ZrO的S-9 号样品400℃时表现出了最高活性,表明,VO和ZrO的共同作用或交互作用进一步提高了催化剂的活性。
由图3 可知,由双变量(图2)改为三变量(图3) 后,各催化剂活性都提高了一个档次(425℃为18%),其中,S-13 催化剂耐热前后的活性都是最高的,说明VO、ZrO、WO助催化剂同时添加对催化剂活性影响较大。对于七助催化剂,S-17 的活性明显高于S-16,表明WO提高活性的作用大于BaO。
2.2 催化剂表征
2.2.1 反应活化能
采用阿伦尼乌斯方程计算了S-1、S-4、S-9、S-13、S-17对反应的表观活化能影响,结果如图4所示。由图4 可知,随着ZrO、VO、WO、MgO助催化剂数量的增加(由1个逐一增加到4个),氨合成反应的表观活化能依次降低。这与催化剂活性的变化规律是完全一致的。

图4 催化剂与活化能的关系
2.2.2 H-TPR分析
图5 是催化剂的H-TPR 曲线,因为所有样品只有一个物相FeO,故只有一个耗H峰,这与文献的结果一致。由图5 可知,与S-1 相比,S-4 的初始还原温度几乎没有改变,但最高峰温向左移15℃,说明ZrO可以促进催化剂还原,这是因为ZrO表面富电子和电子易传递性使得以ZrO为载体的FeO易还原。另外ZrO晶体表面离子具有接受孤对电子的能力,削弱了铁氧离子间的静电吸引力,从而削弱了铁氧键的强度,使H 易夺走FeO中的氧,从而促进了催化剂的还原。在熔铁催化剂中,ZrO主要以CaZrO的形式存在,所以这也是添加ZrO的熔铁催化剂易还原的一个重要原因,由于Ca可以进入ZrO晶格,它具有较高浓度的氧负离子空位,高温下表现出高的导电性,在还原时能起到载流子作用,促使氧化铁中的氧负离子传递,有利于和氢反应,使还原易于进行。

图5 催化剂的H2-TPR还原曲线
S-9、S-13 与S-4 相比,发现催化剂的初始峰温和最高峰温变化不大,说明VO、WO对催化剂的还原性能影响不大。S-17 催化剂的最高峰温明显右移,说明MgO 使催化剂较难还原,这可能是因为AlO、MgO 和Fe 形成了致密难还原的固溶体,降低了还原速度,这也说明了S-17 催化剂耐热前没有被充分还原,故活性较低。
2.2.3 N-TPD分析
对于氨合成催化剂来说,N的脱附活化能与氨合成反应活化能一致。由图6 可知,与S-1 相比,S-4、S-9、S-13 的N脱附峰依次左移,脱附温度降低,脱附活化能减小,有利于氨合成反应的进行。值得注意的是,S-17 的脱附峰温度与S-1 几乎相同。表2 给出了催化剂脱附峰的面积及温度。脱附峰的面积越大,说明其表面的活性位数目越多。图7 关联了N脱附峰的面积与催化剂活性的关系。由图7 可知,S-1、S-4、S-9、S-13、S-17 催化剂的N脱附峰面积与催化剂的活性规律一致。因此,ZrO、VO、WO、MgO 提高了催化剂活性位数目,这可能是催化剂活性提高的重要原因。

图6 各催化剂N2-TPD曲线图

图7 催化剂的N2脱附峰面积及催化剂活性
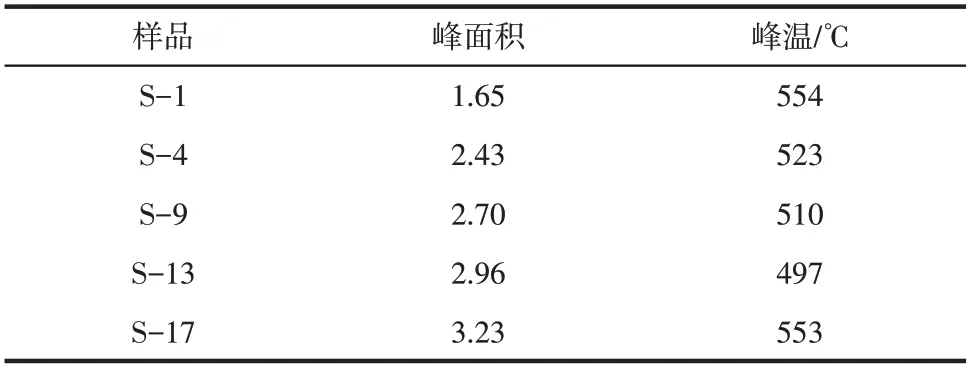
表2 催化剂N2-TPD的参数
2.2.4 XRD分析
图8是S-1、S-4、S-9、S-13、S-17催化剂还原前后的XRD结果。由图8可知,氧化态催化剂都有五个峰,分别在36.09°、41.92°、60.78°、72.77°、76.58°,这归属于维氏体的特征衍射峰。还原后催化剂同样都有五个峰,分别在44.67°、65.02°、82.33°、98.94°、116.38°,这归属于α-Fe的特征衍射峰。没有检测到助催化剂及其可能形成的诸如KAlO、FeAlO、CaZrO和WO等固溶体。这可能是因为助催化剂的含量少,且在催化剂中分布较均匀或仪器精度低所致。

图8 氧化态催化剂和还原态催化剂的XRD
采用谢乐公式计算了不同晶面的晶粒尺寸,见表3。据Somorjai 研究可知,Fe 的不同晶面的氨合成活性大小顺序为(111)>(211)>(100)>(210)>(110),由于对称性原因,X 射线粉末衍射仪不能检测到(111)晶面,故选择检测次高活性的(211)晶面和最低活性的(110)晶面,计算它们的晶粒度比值211/110,该比值代表着两个晶面的相对发育程度,与活性相的活性位数目直接相关。根据晶粒度与晶面发育刚好相反的原则,晶粒度比值越小说明晶面发育越好,活性位越多,即与催化剂活性呈反比。从表3可知,S-1、S-4、S-9 的211/110 比值依次降低,与催化剂活性呈反比,说明催化剂活性提高的一个原因是活性位数目增加,这也与N-TPD 结果一致。但S-13 的211/110比值比S-9高,所以S-13活性应该低于S-9,这与活性结果不一致。这是因为WO加入后会使催化剂中以WO的形式存在,WO晶格上的电子会转移到α-Fe,使α-Fe 上的电子密度增加,相应的电子逸出功减小,从而促进对N的活化吸附,提高催化剂的活性。S-17的211/110比值比S-13小,这与活性结果成反比,这说明S-17催化剂活性提高的原因也是活性位数目增加。
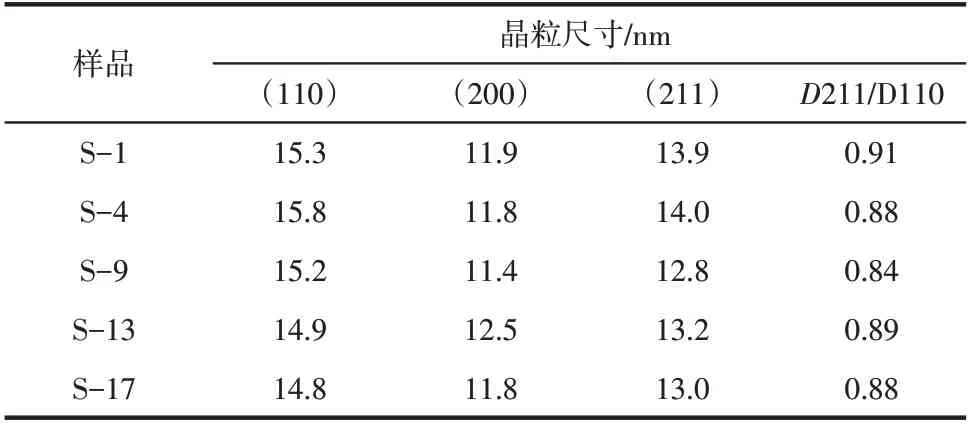
表3 还原态催化剂的晶粒尺寸
2.2.5 EDS分析
图9是S-1、S-4、S-9、S-13、S-17催化剂表面的元素分布。在FeO基氨合成催化剂中,助催化剂的均匀分布也是提高催化剂活性的重要因素。由图9 可知,在S-1 中添加ZrO后催化剂(S-4)表面的Fe、Al、K、Ca、Zr 元素明显比S-1分布均匀,说明ZrO促进了催化剂表面的元素均匀分布。在S-4 中再添加VO(S-9),催化剂表面Al、K、Ca的含量高于S-4(见表4),可能部分元素发生聚集,所以未观察到S-9表面元素分布比S-4更加均匀。但S-9活性则比S-4活性高一个档次(一个百分点),可能VO的主要作用是促进N的吸附,从而提高催化剂活性。S-9 中再添加WO(S-13),明显促进了Zr 和V 元素的均匀分布,且WO在催化剂表面分布均匀。在S-13 中添加MgO(S-17),催化剂表面所有元素都分布得更均匀,这是因为MgO 可以在0~100%范围内与FeO 形成完全互熔体,能够促进所有元素均匀地分布在催化剂母体相中,且S-17表面的Fe含量最高(见表4),说明活性位暴露得较多,所以催化剂活性最高。S-13、S-17 催化剂表面WO含量为零(见表4),可能是WO在催化剂表面分布得较均匀或受仪器精密度的影响。

图9 催化剂EDS表面元素分布图

表4 催化剂表面元素质量分数 单位:%
2.2.6 低温N物理吸附分析
低温N物理吸附实验结果如表5 所示。由表5可知,S-4、S-9 催化剂的比表面积依次增加,孔径减小,这与催化剂的活性变化也一致,这说明ZrO、VO可以使催化剂比表面积增大。值得注意的是,与S-9 催化剂比较,发现S-13、S-17 催化剂的比表面积依次降低,但都高于S-1催化剂的比表面积,这不能说明催化剂比表面积的大小是提高活性的关键因素。

表5 催化剂的低温N2物理吸附实验结果
图10 为各催化剂的还原态FeO 基催化剂N物理吸附脱附等温线和孔径分布图,所有催化剂都表现出Ⅳ型吸附-脱附等温线,具有明显的H型滞后环,这表明催化剂具有不规则的孔结构。孔径分布如图10(b)所示,S-4、S-9、S-13、S-17 催化剂的孔径左移,说明添加ZrO、VO、WO、MgO 助催化剂后催化剂的孔径减小,但是不会改变孔的类型。

图10 催化剂的N2吸附脱附等温线和BJH法计算的孔径分布
3 结论
在AlO、KO、CaO 助催化剂的基础上对MgO、VO、ZrO、WO四种助催化剂进行研究,优选出了四助剂、五助剂、六助剂、七助剂的氨合成FeO 基催化剂最优配方。结果表明,MgO、VO、ZrO、WO都可以提高FeO 基氨合成催化剂的活性,其中ZrO和MgO 有利于催化剂中元素的均匀分布,提高催化剂的活性位数目,促进催化剂的还原,增加催化剂的比表面积,提高了催化剂的耐热性能;VO和WO降低表观活化能、增加α-Fe 上的电子密度,提高催化剂的活性。ZrO和MgO 偏碱性而VO和WO偏酸性,它们之间存在酸碱协同作用。
