宿主细胞转录因子E2 相关因子2 对EIAV 复制影响的初步研究
2022-04-25马官芹林跃智王晓钧
马官芹,王 岩,林跃智,王晓钧
(中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室/马传染病和慢病毒病研究创新团队,黑龙江 哈尔滨 150069)
转录因子E2 相关因子2(Nrf2)属于CNC(Cap-ncollar)转录因子家族成员,是细胞重要的抗氧化因子。阻滞Nrf2 的激活则会加重机体的氧化应激状态,Nrf2 的激活依赖其负调控因子胞浆蛋白kelch 样环氧氯丙烷相关蛋白1(Keap1)。Nrf2 和Keap1 形成复合物以其非活性状态存在于细胞质中,并通过Keap1 介导的泛素化持续降解Nrf2,以保持生理状态下Nrf2 的低水平状态[1]。当发生氧化应激时,机体会通过多种方式诱导或促进Nrf2 和Keap1 的解离,导致Keap1 对Nrf2 的泛素化减弱,Nrf2 蛋白则会发生细胞质到细胞核的移位和磷酸化,启动下游抗氧化反应元件(ARE)靶基因的表达,调控Ⅱ相代谢酶(GST、NQO1 等)、抗氧化酶(SOD、HO-1 过氧化物酶-1 等)的转录活性,从而发挥其抗氧化损伤作用[2]。
宿主细胞Keap1-Nrf2-ARE 通路不仅在病毒感染中起重要作用,其调控病毒在机体内复制的机制也很复杂[3]。病毒对该通路具有双向调控作用:一方面病毒要利用感染造成的氧化应激环境复制,因此要抑制Nrf2-ARE 抗氧化通路的激活。如呼吸道合胞病毒(Respiratory syncytial virus,RSV)可通过蛋白酶体途径诱导Nrf2 的去乙酰化和泛素化[4],牛疱疹病毒1 型(Bovine herpesvirus 1,BoHV-1)可促进Nrf2 的降解和抑制Nrf2 的乙酰化,从而抑制Nrf2 的激活[5];另一方面持续性的氧化应激会致宿主细胞的损伤,已证实有多种病毒需要通过激活Nrf2-ARE 通路来调控氧化与抗氧化的平衡。如人类免疫缺陷病毒(Human immunodeficiencyvirus,HIV-1)通过gp120 和Tat诱导星形胶质细胞中Nrf2-ARE 的激活[6],乙型肝炎病毒(Hepatitis B,HBV)也可通过编码HBx 和膜蛋白LHB 激活该通路[7]。总之,病毒已演化出多种调控Nrf2-ARE 通路的途径和方式来“绑架”宿主的抗氧化进程,以便利于自身的复制。
近年来,CRISPR/Cas9 技术由于具有操作简便,实验周期短等优势在基因组编辑以及治疗癌症、感染和遗传疾病方面显示出巨大潜力。Cas9 和sgRNA是CRISPR/Cas9 系统基因组编辑的重要组成部分:sgRNA 负责定位,Cas9 参与靶位点DNA 的切割。靶位点的识别需要前间隔序列邻近基序(PAM),因此任何含有NGG 基序的DNA 序列均可被识别为靶位点,从而可快速实现对靶基因的敲除、敲入及修饰等。基于此,本研究利用CRISPR/Cas9 技术构建Nrf2基因敲除的HEK293T 细胞系,利用该细胞初步研究了Nrf2 对EIAV 的抗病毒效果,为进一步研究和阐明Nrf2 调控病毒复制的机制研究提供有效工具。
1 材料与方法
1.1 主要实验材料人胚胎肾细胞(HEK293T)由本实验室购买并保存;EIAV 假病毒包装质粒(VSVG、Pony8.1、UKgp)、EIAV 感染性克隆(CMV3-8)、pVR1012-flag-Nrf2 表达载体均由本实验室构建并保存;ExTaqDNA 聚合酶购自Toyobo 公司;限制性内切酶BsmB I 购自Thermo Fisher Scientific 公司;Lenti-CRISPRv2-GFP 载体购自Addgene 公司;RNA 提取试剂盒购自Bio Flux 公司;反转录试剂盒购自TaKaRa公司;PCR 产物纯化试剂盒、DNA 小提试剂盒购自南京诺唯赞生物科技有限公司;TRIzol 试剂购自Invitrogen 公司;CCK-8 细胞活性检测试剂盒购自上海碧云天生物技术有限公司;细胞裂解液、5×蛋白上样缓冲液由本实验室制备;蛋白Marker 购自Thermo Fisher Scientific 公司;DNA 转染试剂PolyjetTM购自Sigma Gen Laboratories 公司;Pen Strep(双抗)购自Gibco 公司;鼠源Nrf2 单克隆抗体(MAb)、EIAV 核蛋白(p26)MAb 由本实验室制备并纯化;兔源Flag标签抗体、兔源actin 标签抗体、鼠源actin 标签抗体、红外荧光标记羊抗兔IgG(IgG-Dylight700)、红外荧光标记羊抗鼠IgG(IgG-Dylight800)、Reverse Transcriptase Assay 试剂均购自Sigma 公司;大肠杆菌DH5α 感受态细胞购自天根生化科技(北京)有限公司。实验中所涉及的引物和测序均由北京擎科新业生物技术有限公司完成。
1.2 sgRNA 的设计根据GenBank 登录的Nrf2 基因序列(S74017.1),利用张锋实验室网站(http://crispr.mit.edu/)靶向Nrf2 基因设计特异性sgRNA,依照sgRNA 的设计原则,选择评分较高的2 组序列作为sgRNA(图1)。
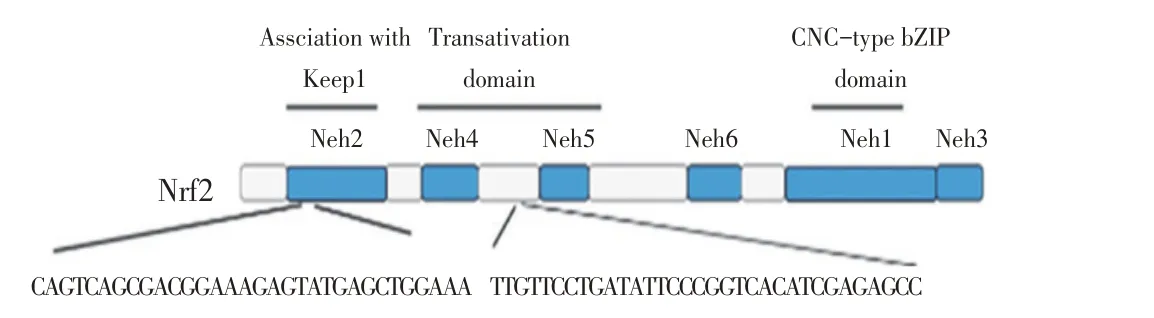
图1 Nrf2基因敲除sgRNA的设计Fig.1 Design strategy of sgRNA for Nrf2 knockout
1.3 LentiCRISPRv2-GFP-sgRNA 质粒的构建与鉴定利用设计的特异性sgRNA(表1),通过程序性退火将两对sgRNA 分别合成双链DNA(98 ℃30 min,95 ℃5 min)后,利用T4 连接酶将该双链DNA 在室温连接至经BsmB I 线性化的载体LentiCRISPRv2-GFP 中,构建重组质粒LentiCRISPRv2-GFP-sgRNAN1 和LentiCRISPRv2-GFP-sgRNA-N2,并分别经测序鉴定后用于敲除细胞系的构建。

表1 靶向Nrf2基因的sgRNA序列Table 1 The sequence of sgRNA targeting Nrf2 gene
1.4 不同sgRNA 沉默Nrf2 基因效率的检测将生长状态良好的HEK293T 细胞铺至6 孔板内,待细胞密度达到80%时,分4 个组利用PolyjetTM转染以下质粒:pVR1012-flag-Nrf2+LentiCRISPRv2-GFP(阴性对照)、pVR1012-flag-Nrf2+LentiCRISPRv2-GFP-sgRNAN1、pVR1012-flag-Nrf2+LentiCRISPRv2-GFP-sgRNAN2 和pVR1012-flag-Nrf2+LentiCRISPRv2-GFP-sgRNAN1+LentiCRISPRv2-GFP-sgRNA-N2。转染24 h 后弃去培养液,PBS 清洗后,每孔加入200 μL 细胞裂解液,10 min 后12 000 r/min 离心5 min,取120 μL 上清加入30 μL 预热的5×loading buffer,98 ℃10 min 后经SDS-PAGE 后。分别以兔源Flag 抗体(1∶5 000)和兔源actin 抗体(1∶10 000)作为一抗,以红外荧光标记羊抗兔IgG(1∶10 000)作为二抗经western blot 检测,并利用Image Studio 软件进行蛋白灰度分析,筛选出沉默Nrf2 基因效率最高的sgRNA。
1.5 Nrf2 基因敲除的HEK293T 细胞系的制备、筛选及鉴定将生长状态良好的HEK293T 细胞铺至6孔板内,待细胞密度达到80%时,利用PolyjetTM试剂转染沉默Nrf2 基因效率最高的LentiCRISPRv2-GFPsgRNA-N1 质粒2 μg,24 h 后收获细胞,利用超速流式分选系统分选出表达GFP 的单一细胞于96 孔板中培养10 d 左右,在显微镜下挑选由单个细胞长成的细胞集落,将其转移至6 孔板内扩大培养。待6 孔板中细胞密度约为80%时,收集细胞,一部分加入细胞裂解液,提取细胞总蛋白(同1.4)后分别利用鼠源Nrf2 MAb(1∶2 000)和兔源actin 抗体(1∶10 000)作为一抗,红外荧光标记羊抗鼠IgG(1∶10 000)、红外荧光标记羊抗兔IgG(1∶10 000)作为二抗经western blot 检测蛋白敲除情况,利用近红外荧光扫描成像系统(Odyssey CLX)结合Image Studio 软件进行蛋白灰度分析。
采用TRIzol 法提取另一部分细胞的总RNA,反转录为cDNA,依据sgRNA 所对应的Nrf2 基因序列位点,设计引物(5′-ATGCAGCCAGATCCCAGGCCTA GCGGGGCT- 3′/5′- ACAGGTACAGTTCTGCTGGTCAAT CTGCTT-3′)经PCR 扩增,PCR 产物经测序鉴定。
1.6 敲除Nrf2 基因后细胞的遗传稳定性检测将经上述方法鉴定为Nrf2 基因敲除的单克隆细胞株(HEK293T-Nrf2KO)传代培养至第10 代(G10)、15 代(G15)、20 代(G20)时分别收取细胞,采用1.5 中的western blot 方法鉴定Nrf2 的表达;按照1×104个/孔分别将HEK293T 和HEK293T-Nrf2KO细胞铺至96 孔板内,设置3 个复孔,待细胞密度达到80%时,每孔加入10 μL CCK-8 溶液,混匀后37 ℃培养1 h,使用酶标仪读取OD450nm处的吸光值,分析Nrf2 敲除后对细胞活力是否产生影响。
1.7 敲除Nrf2 基因对EIAV 复制影响的检测将生长状态良好的HEK293T 和HEK293T-Nrf2KO细胞铺至6 孔板,参照1.5 的方法分别将EIAV 的感染性克隆CMV3-8及Nrf2 基因回补的感染性克隆(CMV3-8+pVR1012-flag-Nrf2)转染至HEK293T-Nrf2KO细胞中,设CMV3-8转染的HEK293T细胞作为对照。转染24 h后收获上清和细胞,分别利用鼠源Nrf2 MAb(1∶2 000)、EIAV 核蛋白(p26)抗体(1∶5 000)、兔源actin 抗体(1∶10 000)以及兔源Flag 抗体(1∶5 000)作为一抗,分别以红外荧光标记的羊抗鼠IgG(1∶10 000)、红外荧光标记的羊抗兔IgG(1∶10 000)作为二抗对上清和细胞中的病毒蛋白经western blot 检测,并对蛋白表达结果经灰度分析EIAV 的复制情况。
1.8 敲除Nrf2 基因后细胞上清中病毒的反转录酶试验(Reverse transcriptase assay,RT)将1.7 中收获的细胞上清各取100 μL 原液、10 倍、20 倍和40 倍稀释液分别与50 μL PEG 8000 混匀,冰浴8 h 后4 ℃,8 000 r/min 离心10 min,弃上清,用40 μL 病毒裂解缓冲液重悬,室温孵育30 min。按照表2 稀释标准品(试剂盒中附带),在重悬的病毒团块和稀释好的标准品中分别加入20 μL 反转录反应混合液(试剂盒中附带),37 ℃孵育1 h。将反转录反应后的样品加入MP 孔(试剂盒中附带)中,另设置60 μL PBS 对照,37 ℃孵育1 h。弃去反应液,用wash buffer 洗涤3 次,每孔加入200 μL地高辛过氧化物酶抗体,37 ℃孵育1 h。弃去反应液,用wash buffer 洗涤3 次,每孔加入200 μL 显色底物,室温孵育5 min~10 min,至出现肉眼可见的绿色。读取OD405nm值,根据标准品的吸光值做出标准曲线,根据标准曲线计算出样品值,以分析培养液上清中病毒粒子的含量。
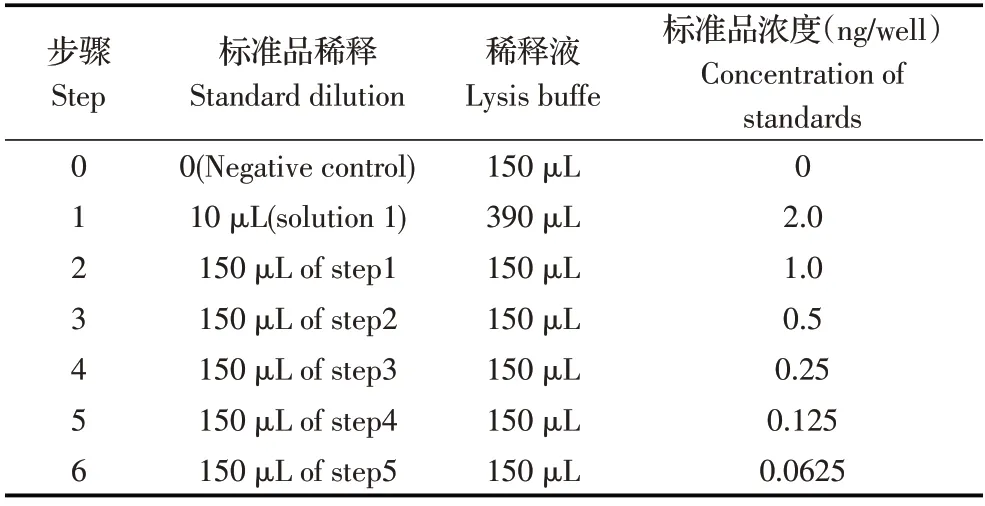
表2 标准品的稀释Table 2 Standard diluent
1.9 EIAV 假病毒感染敲除Nrf2 的细胞后病毒复制水平的检测将包装EIAV 假病毒所需要的质粒UKgp、Pony8.1、VSV-G 按照3∶3∶1 的比例共转染HEK293T 细胞,48 h 收获含有病毒的上清液。将生长状态良好的HEK293T 和HEK293T-Nrf2KO细胞分别铺至24 孔板内,设置3 个复孔,待细胞生长密度达到80%时,加入100 μL 假病毒上清液,24 h 后弃去上清,每孔加100 μL 细胞裂解液,利用微孔板化学发光检测仪检测病毒液萤火虫荧光素酶的活性值,分析EIAV 假病毒在细胞中的复制水平。
2 结 果
2.1 LentiCRISPRv2-sgRNA 基因敲除质粒的构建与鉴定结果将sgRNA-N1 和sgRNA-N2 分别退火获得双链DNA,克隆至LentiCRISPRv2-GFP载体,构建重组质粒LentiCRISPRv2-GFP-sgRNA-N1 和LentiCRISPRv2-GFP-sgRNA-N2,经测序分析结果显示,可检测到正确的sgRNA序列,表明重组质粒正确构建。
2.2 不同sgRNA 沉默Nrf2 基因效率的检测结果将构建的两个LentiCRISPRv2-GFP-sgRNAs 分别与表达Nrf2 的质粒共转染HEK293T 细胞,western blot 检测Nrf2 的表达并进行蛋白灰度值分析。结果显示,与对照组相比,LentiCRISPRv2-GFP-sgRNA-N1 和LentiCRISPRv2-GFP-sgRNA-N2 以及LentiCRISPRv2-GFP-sgRNA-N1+LentiCRISPRv2-GFP-sgRNA-N2 均能显著下调Nrf2 的表达(P<0.01)(图2A、图2B),表明设计的sgRNA均具有较高的沉默效率,本研究选择LentiCRISPRv2-GFP-sgRNA-N1质粒用于后续实验。
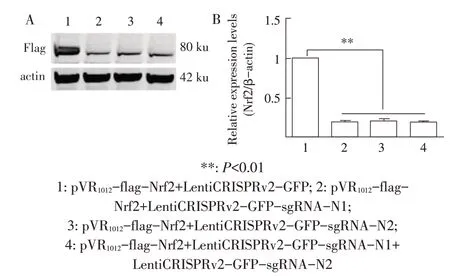
图2 sgRNA敲除Nrf2基因的western blot检测(A)和灰度值分析(B)Fig.2 Western blot detection(A)and gray value analysis(B)of sgRNA knockout Nrf2 gene
2.3 Nrf2 基因敲除的HEK293T 细胞系的构建与筛选将LentiCRISPRv2-GFP-sgRNA-N1 转染HEK293T细胞,通过流式细胞仪和有限稀释法筛选表达GFP的单细胞克隆。经过10 d 培养,将单克隆细胞长成的细胞集落经western blot 鉴定。结果显示,N2 和N4 2 株单细胞克隆株未检测到Nrf2 的内源性表达(图3A)。随后提取2 株细胞总RNA,PCR 扩增后测序鉴定,结果显示,Nrf2基因敲除的细胞基因组缺失一个碱基T,导致Nrf2 基因发生移码突变无法表达(图3B)。表明获得了Nrf2基因敲除的HEK293T 细胞。
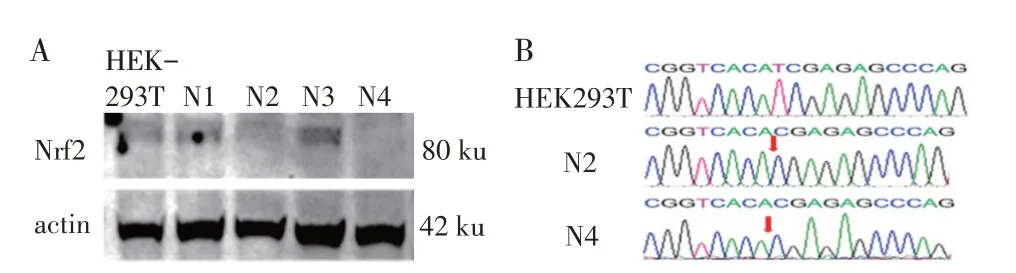
图3 HEK293T-Nrf2KO细胞中Nrf2基因表达的western blot(A)和测序(B)鉴定结果Fig.3 Western blot(A)and sequencing(B)identification of Nrf2 gene expression in HEK293T-Nrf2KO cells
2.4 敲除Nrf2 基因后细胞的遗传稳定性检测结果将获得的HEK293T-Nrf2KO细胞传代,并随机选取G10、G15、G20 代次的细胞经western blot 检测Nrf2的表达。结果显示,3 个代次细胞中均未检测到内源性Nrf2 的表达(图4A、图4B)。利用CCK-8 试剂盒检测HEK293T-Nrf2KO细胞的活性,结果显示,该细胞与HEK293T 具有相似的生长活性,即Nrf2 基因的敲除未对细胞活性产生影响(图4C)。以上结果表明获得了Nrf2 基因敲除的稳定细胞系。

图4 Nrf2基因敲除细胞系的遗传稳定性的western blot(A)及其灰度值分析(B)和CCK-8法检测细胞的活性(C)Fig.4 Genetic stability of Nrf2 knockout cell lines by western blot(A)and its gray value analysis(B)and CCK-8 assay for cell activity(C)
2.5 敲除Nrf2 基因对EIAV 增殖影响的检测结果为了研究所获得的Nrf2 敲除细胞系是否影响EIAV的复制,将EIAV 的感染性克隆CMV3-8及Nrf2 基因回补的感染性克隆(CMV3-8+pVR1012-flag-Nrf2)分别转染至HEK293T-Nrf2KO细胞中,并设置转染CMV3-8的HEK293T 细胞作为对照。通过western blot 检测各组细胞中病毒蛋白Gag(剪切体为p55、p26,p26 可分泌至细胞外)和上清中病毒蛋白(p26)的表达水平。结果显示,与转染CMV3-8的HEK293T 细胞相比,转染CMV3-8的HEK293T-Nrf2KO细胞与上清中病毒蛋白Gag 的表达量均上调约1.5 倍,而转染CMV3-8+pVR1012-flag-Nrf2 的HEK293T-Nrf2KO细胞与上清中病毒蛋白Gag 的表达量均下调约50%(图5A、图5B),可见敲除Nrf2 基因促进了细胞和上清中Gag 蛋白的表达(本实验室制备的p26抗体可同时检测到Gag两个剪切体p55、p26)。表明Nrf2 基因可以抑制EIAV 在HEK293T细胞中的表达。

图5 敲除Nrf2对EIAV增殖影响的western blot检测结果(A)及其灰度值分析(B)Fig.5 Detection results of the effect of Nrf2 knockout on EIAV proliferation(A)and analysis of its grayscale values(B)
2.6 敲除Nrf2 基因后细胞上清中病毒的RT 结果将收获的上述各组细胞上清各取原液及不同倍数的稀释液分别与50 μL PEG8000 混匀、离心、裂解后,利用RT检测其中病毒反转录酶的含量,以分析各组培养液上清中病毒粒子的含量。结果显示,与转染CMV3-8的HEK293T细胞相比,转染CMV3-8的HEK293T-Nrf2KO细胞上清中病毒逆转录酶的含量上调约1.5倍,而转染CMV3-8+pVR1012-flag-Nrf2 的HEK293T-Nrf2KO细胞上清中的病毒逆转录酶含量下调约50%(图6)。再次证明Nrf2 基因具有抑制EIAV 复制的作用。
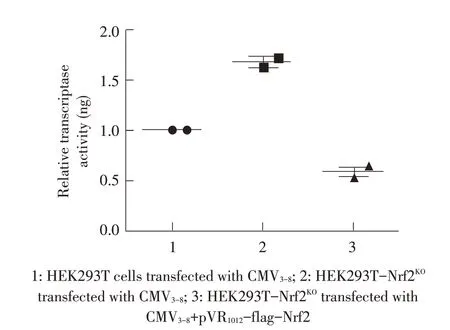
图6 敲除Nrf2基因后细胞中病毒复制能力的检测结果Fig.6 Detection of viral replication capacity in cells after knockout of Nrf2
2.7 EIAV假病毒感染Nrf2基因敲除细胞后的病毒复制水平的检测结果为进一步探究Nrf2 调控EIAV 复制的分子机制,本研究利用重组质粒UKgp、Pony8.1、VSV-G 共转染HEK293T 细胞制备EIAV 假病毒,将其分别感染HEK293T 和HEK293T-Nrf2KO细胞,24 h 后裂解细胞通过检测萤火虫荧光素酶的活性,评价该假病毒在细胞内的复制水平。结果显示,假病毒感染的HEK293T-Nrf2KO细胞上清中荧光素酶的活性与其转染的HEK293T 细胞相比上调约2.5 倍(P<0.01)(图7),进一步表明Nrf2 抑制了EIAV 在HEK293T 细胞中的复制。

图7 EIAV假病毒感染敲除Nrf2细胞后的病毒复制水平检测结果Fig.7 Detection of viral replication levels after Nrf2 knockout cells infection with EIAV pseudovirus
3 讨 论
CRISPR/Cas9 系统应用最广泛,且具有设计简单、易于使用和多路复用(能够同时编辑多个基因)的优点,同时,它还能够有效地修饰各物种和不同细胞类型的内源性基因[8-9]。另外,带有转录抑制因子或激活因子的改良CRISPR/Cas9 系统可以强有力的转录抑制或激活靶基因[10]。但在长期的应用中发现CRISPR/Cas9 技术经常发生脱靶的问题[11],所以本研究在进行CRISPR/Cas9 试验时,利用NCBI 检索了Nrf2 全基因,之后利用张峰实验室网站靶向Nrf2第一和第二外显子分别设计了sgRNA。试验结果表明,本研究所设计的两条sgRNA的切割效率均高效。
Nrf2-ARE 作为宿主抗氧化防御的核心途径,在调控宿主的抗氧化损伤过程中发挥重要作用,并参与肿瘤、凋亡、自噬和炎症等多种病理过程的调控。目前,主要研究的是Nrf2的抗病毒功能,而对其抗病毒机制研究的较少。本研究利用CRISPR/Cas9 系统,设计了2 条靶向Nrf2 的sgRNAs,将sgRNAs 插入LentiCRISPRv2-GFP 载体,获得Nrf2 基因敲除的重组质粒。将重组质粒转染HEK293T 细胞,通过流式筛选和无限稀释的方法获得Nrf2 基因敲除的HEK293T 细胞系HEK293T-Nrf2KO细胞,通过多次传代并鉴定后确定该细胞系可稳定遗传。本研究将EIAV 的感染性克隆CMV3-8,及EIAV 的假病毒分别感染该细胞系,通过western blot、RT及荧光素酶活性检测表明Nrf2蛋白能够抑制EIAV在细胞内的复制。
Nrf2-ARE 是宿主细胞抗氧化防御体系的第一道防线,是其对抗病毒感染引起氧化应激最重要的防御系统。许多具有抗氧化防御功能的蛋白质,都依赖于Nrf2 通路的转录调控。Nrf2-ARE 通路对宿主细胞氧化还原平衡的维持、氧化应激致细胞损伤的预防及炎症平衡的调节机制是复杂多样的。而机体内氧化/抗氧化这一平衡的移动方向直接关系到病毒所致疾病的发展方向。因此,对病毒和宿主Nrf2 通路调控机制的深入认知,明确如何调控抗氧化途径治疗病毒性疾病,成为氧化应激相关疾病(病毒性疾病、癌症、心血管系统疾病和炎症等)预防和治疗的热点。本研究团队还利用该细胞系检测了敲除Nrf2 基因对马流感病毒复制的影响,获得的结果与本研究结果一致(数据未显示),推测Nrf2 对病毒在细胞中复制的抑制作用可能具有普遍性。本研究构建的HEK293T-Nrf2KO细胞系可为Nrf2 基因的抗病毒机制研究提供重要的技术平台,也可为Nrf2-ARE 通路调控病毒机制的研究奠定物质基础。
