伊曲康唑胶囊的质量评价
2022-04-20杜亚俊张秉华牛龙青衷红梅赵蒲中
杜亚俊 张秉华 牛龙青 衷红梅 赵蒲中
(陕西省食品药品检验研究院,西安 710065)
伊曲康唑为三唑类抗真菌药物,1980年由比利时杨森制药有限公司(Janssen Pharmaceutica N.V.)合成。伊曲康唑通过干扰真菌细胞膜麦角固醇的生物合成来达到抑制真菌增殖、促进真菌死亡的目的,具有抗菌谱广、毒性低的特点,临床上适用于浅表真菌感染和深部真菌引起的系统感染,还适用于其他抗真菌药物不适用或无效的系统性感染[1-3]。1987年伊曲康唑胶囊首次在墨西哥获准上市,1993年在我国获准进口。2000年,伊曲康唑胶囊在国内天津力生制药厂首仿获准生产。目前国内有批准文号5个,生产企业5家,规格均为0.1 g。其不良反应主要表现为胃肠道不适、肝功能异常和头痛。
2015年在国家药品抽验计划中,笔者曾对伊曲康唑胶囊进行过质量评价,2021年伊曲康唑胶囊再次作为国家药品抽检计划品种。根据标准查询情况,现行的中国药典(ChP)、美国药典(USP)、欧洲药典(EP)、英国药典(BP)及日本药局方(JP)均收载了伊曲康唑原料,ChP、USP及企业注册标准YBH12752020收载了伊曲康唑胶囊。本文按现行质量标准对全部样品进行检验,并针对其中发现的问题开展相应的探索性研究,通过对关键指标与2015年国家药品抽验计划结果的对比,对当前伊曲康唑胶囊的质量状况进行客观评价,并为进一步完善质量标准提出建议。
1 仪器及试药
1.1 仪器与试剂
高效液相色谱仪(Agilent 1260及岛津 LC-2030 3D),电子天平(瑞士梅特勒XPE205及ME204),气相色谱仪(岛津GC-2010 Plus及Agilent 8890),溶出度仪(Agilent 708-DS、天大天发RC806ADK、天大天发RC1208D及SOTAX AT 7x),紫外可见分光光度计(岛津UV 2600),水分活度仪(AquaLab 4TE,美国METER Group, Inc.公司)
四氢呋喃(色谱纯,Merck公司),甲醇、乙腈(色谱纯,美国Sigma-Alorich公司),四丁基硫酸氢铵、盐酸、乙酸铵、十二烷基硫酸钠(分析纯,国药集团)
1.2 对照品
伊曲康唑对照品(批号:100631-201402,含量:99.2%)、杂质A对照品(批号:130686-201501,含量:99.5%)、杂质B对照品(批号:130687-201501,含量:97.5%)、杂质C对照品(批号:130688-201501,含量:96.3%)、杂质D对照品(批号:130689-201501,含量:95.1%)、杂质E对照品(批号:130690-201501,含量:97.1%)、杂质F对照品(批号:130691-201501,含量:98.0%)、杂质G对照品(批号:130692-201501,含量:95.5%),均购自中国食品药品检定研究院。杂质H对照品(杂质名称自拟,企业D提供,批号:WVLA_0062_003_1,含量:97.2%)。
1.3 样品
伊曲康唑胶囊为2021年国家评价性抽验样品,共131批,抽自全国29个省、自治区和直辖市的药品生产、经营和医疗单位,样品涉及A、B、C和D 4个生产企业。企业A、B和C均提供了原辅料;企业D采用从国外进口伊曲康唑小丸,在本地进行胶囊填充和泡罩包装生产伊曲康唑胶囊,未提供原辅料。企业D的产品为原研药品地产化品种,于2018年经国家食品药品监督管理总局仿制药质量与疗效一致性评价专家委员会审核确定为仿制药参比制剂。
2 实验方法
2.1 法定检验
检验依据为ChP 2015年版第一增补本和2020年版二部,主要项目包括性状、 鉴别、有关物质、二氯甲烷、溶出度、装量差异和含量测定,两个标准内容一致。
2.2 探索性研究
2.2.1 杂质谱分析
采用现行标准有关物质检查的方法,通过杂质对照品定位和加速试验,对原料药生产过程可能引入的工艺杂质、制剂生产及贮存过程中可能产生的降解产物进行杂质归属,明确各企业样品的关键杂质,比较分析国产制剂与原研地产化药品杂质水平的差异。
2.2.2 有关物质测定方法研究
针对部分样品相邻杂质峰分离效果不佳的问题,建立了新的方法测定有关物质。色谱条件:色谱柱:Aglient Proshell 120 EC-C18(150 mm×4.6 mm,2.7 μm);柱温:30℃;检测波长:260 nm;流速:0.8 mL/min;进样量:10 μL;流动相A:0.01 mol/L乙酸铵溶液;流动相B:乙腈,梯度洗脱(表1);稀释剂Ⅰ:甲醇-乙腈(3:1)、稀释剂Ⅱ:量取稀释剂Ⅰ700 mL,加0.01 mol/L乙酸铵溶液300 mL,混匀。采用该方法对每个企业3批样品进行测定。
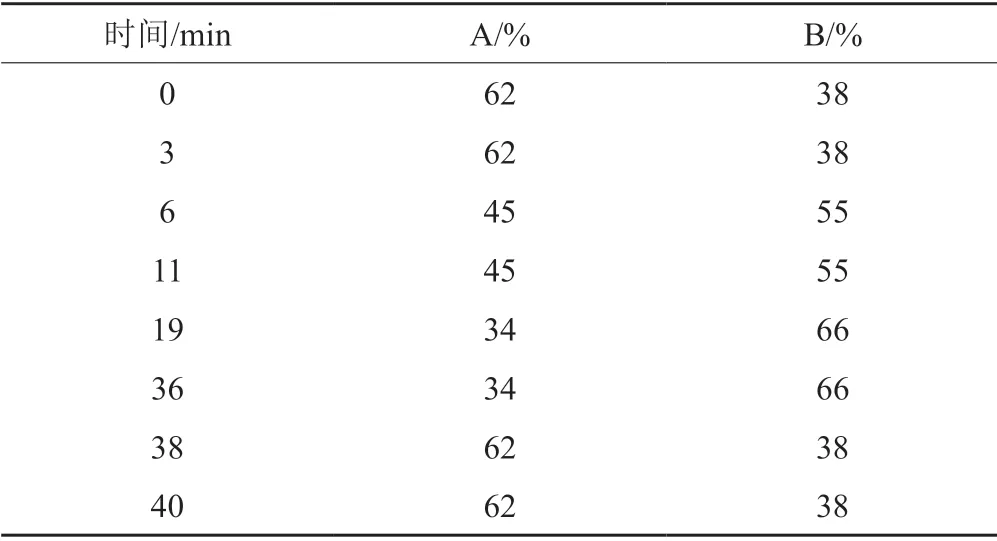
表1 HPLC法流动相梯度洗脱表Tab.1 The gradient elution table of HPLC mobile phase
2.2.3 溶出行为及溶出度方法考察
伊曲康唑为难溶性弱碱性药物,溶解性跟溶剂的pH紧密相关。查询日本橙皮书发现,伊曲康唑胶囊(规格50 mg)在pH4.0醋酸盐缓冲液、pH6.8磷酸盐缓冲液和水3种介质中,桨法,转速50 r/min条件下,6 h的溶出量均在5%以下;查阅文献[4]发现,企业B、D和C的伊曲康唑胶囊在pH4.0醋酸盐缓冲液、pH6.8磷酸盐缓冲液和水3种介质中,桨法,转速100 r/min条件下,1.5 h的累积溶出量在5%以下,溶出曲线无区分度,因此不再考虑这3种溶出介质。探索性研究中,按照《普通口服固体制剂溶出度试验技术指导原则》要求,选择已形成标准的3种方法绘制溶出曲线,对溶出数据进行比较以选择出较为科学合理的溶出度方法,同时比较国产制剂与参比制剂的相似性。
方法1:参照现行的中国药典伊曲康唑胶囊溶出度方法[5],采用桨法,以盐酸溶液(9→1000)1000 mL为溶出介质,转速为75 r/min。
方法2:参照比利时杨森公司(Jassen Pharmaceutica N.V.)的伊曲康唑胶囊全球释放标准中溶出度方法,采用桨法,以含1%吐温-20的盐酸(7→1000)氯化钠溶液900 mL为溶出介质,转速为100 r/min。
方法3:参照美国药典伊曲康唑胶囊溶出度方法1[6],采用桨法,以含0.25%(W/V)十二烷基硫酸钠的盐酸溶液(9→1000)900 mL为溶出介质,使用沉降篮,转速为75 r/min。
2.2.4 稳定性考察
按照现行的中国药典9001原料药物与制剂稳定性试验指导原则[7],每个企业取3批样品按市售包装置于温度40℃±2℃、相对湿度75%±5%的条件下,分别在第1、2和3月末取样一次,拍照记录性状变化,同时测定有关物质。
2.2.5 水分活度测定
水分活度(water activity)可以表征口服固体制剂产品中微生物可利用的水分情况,是影响微生物生长的关键因素之一,水分活度可反映产品中潜在微生物的生长繁殖状况[8],探索性研究中,采用仪器测定法对4个企业的样品水分活度进行了测定,为非无菌制剂微生物污染风险评估提供参考。
3 结果与讨论
3.1 法定检验结果
按现行质量标准检验,131批样品均符合规定,合格率为100%,较2015年国评合格率(94.7%)[9]有明显提升。结合探索性研究结果,对产品质量密切相关且各企业产品间差异较大的项目(有关物质和溶出度)进一步进行比较分析。
3.2 杂质谱比较
对131批伊曲康唑胶囊进行检验,典型色谱图见图1,发现主要检出7个杂质,利用中国食品药品检验研究院提供的伊曲康唑杂质A~G对照品及企业提供的杂质H对照品定位杂质峰,确定了其中5个已知杂质,分别为杂质B(编号1),杂质C和杂质D(编号2),杂质F(编号3),杂质H(编号5),杂质G(编号6),编号4和7的杂质为未知杂质。
所有批次样品按现行标准检验, 企业A主要检出3~4个杂质,分别为杂质B、杂质C+D、杂质F和编号为4的未知杂质;最大单个杂质为杂质B,分布在0.07%~0.2%之间,杂质总量分布在0.13%~0.45%之间;企业B主要检出4个杂质,分别为杂质B、杂质C+D、杂质G和杂质H,最大单个杂质为杂质B或杂质H,分布在0.09%~0.3%之间,杂质总量分布在0.16%~0.55%之间;企业C主要检出4~5个杂质,分别为杂质B、杂质C+D、杂质G和编号为7的未知杂质,最大单个杂质为杂质B,分布在0.09%~0.2%之间,杂质总量分布在0.11%~0.51%之间。企业D主要检出2个杂质,分别为杂质B和杂质E,多批次样品杂质检出量在忽略限以下,最大单个杂质分布在未检出~0.1%之间,杂质总量分布在未检出~0.25%之间。企业A、B、C的产品中杂质个数与杂质含量均高于企业D的产品,说明国内生产企业的产品在有关物质方面与原研地产化药品仍然存在一定差距。各企业样品最大单个杂质和杂质总量箱式图见图2。
通过对企业A、B、C提供的伊曲康唑原料进行检测发现,企业A、C的原料中未检出杂质H,该杂质在胶囊剂中未检出或含量多在忽略限(约0.05%)以下;企业B的原料中检出杂质H,但含量明显低于制剂;其余检出杂质在原料和制剂中含量基本相当。结合加热(105℃)破坏试验和加速试验结果可知,杂质H为热降解杂质,制剂生产过程可能由干燥环节降解产生。综上可知,胶囊剂中的杂质水平基本由其原料药的杂质水平决定,提示伊曲康唑胶囊中的杂质主要来源于原料药,控制好原料药生产工艺是保证制剂质量的关键。从制剂有关物质检验情况推测,原研地产化产品由原料引入的工艺杂质较低。
将此次杂质谱分析结果与2015年国抽结果进行比较,发现各企业杂质检出个数及杂质总量基本一致。2015年国抽中,企业A的样品和原料中未检出编号为4的未知杂质,推测企业A杂质谱变化由更换原料引起。
采用优化的HPLC方法对诸企业样品有关物质进行测定。检验结果显示,企业A的样品杂质总量结果高于现行标准方法,企业B、C、D的样品检测结果与现行标准方法测定结果基本一致。
3.3 溶出度比较
131批样品溶出度均值分布在83%~105%之间,绘制溶出度均值频数分布图(图3),可见结果呈正态分布,溶出度限度设置合理。
3.3.1 不同企业产品的溶出度/溶出行为差异
比较各企业样品的批内溶出度结果,发现企业D的产品个别批次单粒溶出度小于80%,有2批样品的批内溶出度RSD大于10%,5批样品的批内溶出度RSD处于5%~10%之间,与企业A、B、C的产品相比,批内差异性较大,RSD散点图见图4。分析其可能原因,企业D的处方中主药成分与辅料的配比较低,使得主药成分在溶出介质中释放较慢。我们注意到,在2015年国家药品抽验中,伊曲康唑胶囊检验标准为ChP 2010年版二部,溶出度检查取样时间为45 min,企业D有2批样品溶出度检查不符合规定,其余批次样品的溶出度批内差异也较大[9]。在ChP 2015年版第一增补本和2020年版二部中,伊曲康唑胶囊溶出度检查将取样时间由45 min延长至60 min,企业D的产品溶出度检查合格率100%,但批内的溶出结果差异仍然较大。鉴于企业D产品采用特殊的微丸制备工艺及添装工艺,且其伊曲康唑胶囊已被确定为参比制剂,提示目前Chp的溶出度测定方法可能并不能完全客观评价该工艺产品的质量。
采用方法1、方法2、方法3绘制溶出曲线(图5)。对溶出数据进行比较,发现方法1中企业D的3批样品在第1个取样点(15 min)的溶出量的RSD均大于20%,在第二个取样点的(30 min)累计溶出量的RSD均大于10%;企业A、B、C的9批样品中有3批样品在第1个取样点(15 min)的溶出量的RSD大于20%,有1批样品在第2个取样点的(30 min)累计溶出量的RSD大于10%。方法2中企业D的3批样品仅1批样品在第2个取样点的(20 min)累计溶出量的RSD大于10%;企业A的3批样品在第1个取样点(10 min)的溶出量的RSD均大于20%,企业B、C各有1批样品在第2个取样点的(20 min)累计溶出量的RSD大于10%。方法3中4个企业12批样品在第1个取样点(10 min)的溶出量的RSD均小于20%,在第二个取样点的(20 min)累计溶出量的RSD均小于10%。上述结果提示,使用沉降篮和在介质中加入适宜适量的表面活性剂可明显降低批内差异,根据不同制剂处方工艺的特点,选择适宜量的表面活性剂是建立合理溶出度检测方法的关键。
按《普通口服固体制剂溶出度试验技术指导原则》要求,采用非模型依赖多变量置信区间法和f2相似因子法比较国内3个生产企业的样品与参比制剂(企业D)的差异。结果显示,企业A样品的溶出曲线与参比制剂均不具有相似性;企业B、C的6批样品在3种方法下的溶出曲线不能同时与参比制剂具有相似性。总的来说,企业A、B、C的产品尤其是企业A产品的溶出偏快,与原研地产化药品存在差异。
3.3.2 溶出度分析方法合理性探讨
溶出度试验是模拟药物体内释放过程的理想体外方法。伊曲康唑为难溶性弱碱性药物,其制剂的工艺与处方决定药物体内的释放过程,如何建立体内外相关的溶出度测定方法是其难点。本文的结果提示,采用相同的溶出度方法评价不同工艺、处方的伊曲康唑胶囊质量具有一定的风险,针对具体产品工艺、处方的差异制订不同的溶出度方法势在必行。
3.4 稳定性考察
稳定性实验中,各企业样品性状变化存在较大差异,见图6。企业A的样品在1个月后内容物小丸表面即有少量粉末脱落;2个月后小丸表面有大量粉末脱落,小丸结构破坏严重。企业C的样品在2个月后小丸表面开始失去光泽,有轻微粉末脱落;3个月后小丸变化与2个月基本一致。企业B和D的样品在3个月后内容物小丸结构依然完整,未发现表面有粉末脱落,但出现个别小丸黏连的现象。有关物质检查结果显示,企业A的样品中杂质H由未检出增大至0.3%左右;企业C的样品中该杂质由未检出增大至0.2%左右;企业B的样品中该杂质由0.1%左右增大至0.3%左右;企业D的样品该杂质由未检出增大至0.09%,增速最缓。从性状和杂质变化情况可以看出,各企业样品内容物小丸稳定性由小到大依次为:企业A<C<B<D,可以看出各企业的处方与生产工艺存在较大差异。提示仿制药一致性评价中,产品处方和工艺直接影响其与参比制剂的一致性。
3.5 水分活度比较
对4个企业的102批伊曲康唑胶囊的水分活度进行测定,每批样品平行测定3份,3次测定结果显示,温度的变化会引起水分活度结果的变化,为减小温度的影响,尽量选取接近25.25℃时测定的水分活度结果绘制伊曲康唑胶囊温度-水分活度簇状柱形图(图7)。由图可知,4个生产企业的产品水分活度均低于0.60,较低的水分活度限制了潜在微生物的生长繁殖,提示该产品微生物污染风险较小。另外还可以看出,在较小的温度范围内,企业D的产品水分活度结果维持在0.4左右,一定程度上反映了该企业生产工艺的稳定性,其余3个企业的产品水活度波动相对较大。
4 总结
按法定标准检验,伊曲康唑胶囊合格率为100%,较2015年国评合格率94.7%[9]有明显提升,结合探索性研究情况认为,伊曲康唑胶囊总体质量情况较好。探索性研究结果显示,伊曲康唑胶囊内容物小丸的稳定性与其溶出行为存在一定的相关性,稳定性差的溶出速度快。国内3个生产企业与原研地产化药品的溶出行为均存在不同程度的差异,在仿制药质量与疗效的一致性评价工作中,应对处方工艺的合理性进行分析,以保证药品疗效。此外,现行的质量标准有关物质和溶出度检查方法存在不足,需进一步完善。
