Box-Behnken 响应面法结合指纹图谱优化决明子总蒽醌提取工艺
2022-04-20邹纯才鄢海燕
王 恒,邹纯才,鄢海燕
(皖南医学院药学院,安徽 芜湖 241002)
决明子为豆科植物决明Cassia obtusifoliaL.或小决明Cassia toraL. 的干燥成熟种子,其味甘、苦、咸,性微寒,善入肝、肾、大肠经,具有清热明目、润肠通便的功效[1-3]。决明子最早记录于《神农本草经》,在我国有悠久的使用历史,也曾被原国家卫生部列为药食同源的中药之一[4],在医疗保健行业有着巨大的应用潜力。现代药理学研究表明,决明子具有降血压、降血脂、抗血小板聚集、免疫、保肝、明目、抗氧化、抑菌等药理作用[5,6],在临床上有着广泛的应用,是中医药治疗心脑血管疾病、高脂血症、便秘的代表性药物[7]。决明子含有蒽醌类、萘并吡喃酮类、脂肪酸类、多糖和氨基酸等许多成分,主要成分是蒽醌类化合物,约占1%[7-9],同时也是决明子中发挥药效的主要物质,有着保肝、抗氧化、抗突变等多种药理活性[10],受到药学相关人员的重点关注。
《中国药典》2020 年版一部对决明子蒽醌类成分如大黄酚、橙黄决明素的含量测定,是以甲醇为提取溶剂提取蒽醌类成分,并以稀盐酸水解结合型蒽醌获得总蒽醌,再以无水乙醇-乙酸乙酯(2∶1)为定容溶剂并进行含量测定。分析提取溶剂的价格、毒性及生产中大量提取的使用等情况,考虑决明子中游离蒽醌和结合型蒽醌共存的实际,结合《中国药典》2020 年版以无水乙醇-乙酸乙酯(2∶1)为水解后游离蒽醌类化合物的定量分析定容溶剂。本研究拟基于响应面设计法结合中药指纹图谱技术考察不同浓度乙醇与乙酸乙酯(2∶1)混合溶剂对决明子总蒽醌化合物提取工艺的影响,明确决明子总蒽醌的最佳提取工艺参数,为后期决明子总蒽醌药理药效学的研究奠定药效成分基础。
1 材料与方法
1.1 仪器
FA2004B 电子天平(上海越平科学仪器有限公司);岛津UV-20A 高效液相色谱仪(岛津公司);DZF-6090 型真空干燥箱(上海三发科学仪器有限公司);Heidolph-LR4010/4011 旋转蒸发仪(德国海道尔夫公司);DF-I 集热式磁力加热搅拌器(常州荣华仪器制造有限公司);KQ-250DE 型数控超声波清洗器(昆山市超声仪器有限公司);RHP-400A 型高速多功能粉碎机(浙江永康市荣浩工贸有限公司)。
1.2 材料
决明子(清炒品,北京同仁堂诚安药材有限公司,批 号:2019073103、2020011901);橙黄决 明 素(成都普菲德生物技术有限公司,纯度:≥98%,批号:17032405);大黄素(中药固体制剂制造技术国家工程研究中心,≥98%,批号:D03-140210);大黄酸(中药固体制剂制造技术国家工程研究中心,纯度:≥98%,批号:D05-140210);大黄酚(成都普菲德生物技术有限公司,纯度:≥98%,批号:17032204);大黄素甲醚(中药固体制剂制造技术国家工程研究中心,纯度≥98%,批号:D04-140210);无水乙醇(安徽安特食品股份有限公司,批号:210307363604)、乙酸乙酯(上海展云化工有限公司,批号:20210220)均为分析纯;甲醇(天津市科密欧化学试剂有限公司,批号:20160129)、乙腈(江苏永华化学科技有限公司,批号:20151102)为色谱纯,水为去离子水。
1.3 方法
1.3.1 溶液的制备
1.3.1.1 决明子总蒽醌提取物的制备 取决明子粉末(过三号筛)约5.0 g,精密称定,置250 mL 圆底烧瓶中,按一定液料比加入一定浓度乙醇与乙酸乙酯按2∶1(mL∶mL)混合的溶液,水浴回流提取3 次,每次至规定时间,抽滤,合并滤液减压浓缩至一定体积,用60%乙醇洗出,水浴挥干溶剂,真空干燥(压力−0.1 MPa,温度70 ℃,下同)48 h,得决明子总蒽醌提取物。
1.3.1.2 决明子总蒽醌供试品溶液的制备 取“1.3.1.1”项下决明子总蒽醌提取物约0.5 g,精密称定,加入稀盐酸30 mL,水浴回流水解1 h,立即冷却,用二氯甲烷萃取5 次,每次30 mL,合并二氯甲烷液,回收溶剂,残渣用无水乙醇-乙酸乙酯(2∶1)混合溶液洗出,转移至25 mL 量瓶中,并用无水乙醇-乙酸乙酯(2∶1)混合溶液定容至刻度,得决明子总蒽醌供试品溶液,备用。
1.3.1.3 决明子总蒽醌参照溶液的制备 按《中国药典》[1]制备决明子总蒽醌参照溶液。取决明子粉末(过三号筛)约5.0 g,精密称定,精密加入甲醇500 mL,加热回流2 h,放冷,抽滤,旋蒸至干,真空干燥,得决明子总蒽醌提取物(参照品)。取决明子总蒽醌提取物(参照品)0.5 g,精密称定,加稀盐酸30 mL,水浴水解1 h,立即冷却,用二氯甲烷萃取5次,每次30 mL,合并二氯甲烷液,回收溶剂,残渣用无水乙醇-乙酸乙酯(2∶1)混合溶液使溶解,转移至25 mL 量瓶中,并用无水乙醇-乙酸乙酯(2∶1)混合溶液定容至刻度,得决明子总蒽醌参照品溶液,备用。
1.3.1.4 对照品溶液的制备 取橙黄决明素、大黄素甲醚、大黄酚、大黄素、大黄酸5 种对照品适量,精密称定,分别置于量瓶中,用无水乙醇-乙酸乙酯(2∶1)配制成质量浓度均为200 μg/mL 的对照品贮备液。再分别精密吸取5 种对照品贮备液适量,置于量瓶中,用无水乙醇-乙酸乙酯配制成质量浓度为12 μg/mL 的混合对照品溶液。
1.3.2 指纹图谱的建立
1.3.2.1 色谱条件[11]色谱柱YMC-Pack ODS-A C18(250.0 mm×4. 6 mm,5 μm);流动相为A(乙腈)-B(0.1%磷酸),梯度洗脱,分析时间100 min;0~15 min,65%~65% B;15~18 min,65%~68%B;18~23 min,68%~68% B;23~25 min,68%~65% B;25~45 min,65%~50% B;45~63 min,50%~45% B;63~85 min,45%~45% B;85~100 min,45%~10% B;柱温为30 ℃;体积流量:1 mL/min;检测波长287 nm。
1.3.2.2 精密度试验 取决明子粉末5.0 g,精密称定,按“1.3.1.2”项下方法制备供试品溶液,按“1.3.2.1”项下色谱条件连续进样5 次,记录色谱图。求得橙黄决明素、大黄酸、大黄素、大黄酚、大黄素甲醚保留时间的RSD 值依次为0.026%、0.033%、0.012%、0.017%、0.023%,峰面积的RSD 值分别为0.14%、0.081%、0.54%、0.24%、0.64%,说明方法精密度良好。
1.3.2.3 重复性试验 取同一批决明子粉末5 份,每份5.0 g,精密称定,按“1.3.1.2”项下方法平行制备供试品溶液,按“1.3.2.1”项下色谱条件进样,记录色谱图。结果显示橙黄决明素、大黄酸、大黄素、大黄酚、大黄素甲醚保留时间的相对标准偏差(rela‑tive standard deviation,RSD)值 依 次 为1.10%、1.30%、0.49%、0.68%、0.78%,峰面积的RSD 值分别为3.4%、3.2%、3.4%、3.0%、2.6%,表明该方法重复性较好。
1.3.2.4 稳定性试验 取同一份供试品溶液,按“1.3.2.1”项下色谱条件,分别于0、3、6、9、12 h 进样分析,记录色谱图。结果显示橙黄决明素、大黄酸、大黄素、大黄酚、大黄素甲醚保留时间的RSD 值依次为0.038%、0.047%、0.110%、0.014%、0.200%,峰面积的RSD 值分别为0.39%、0.97%、0.61%、0.22%、0.72%,表明供试品溶液在12 h 内稳定。
1.3.3 单因素试验 按“1.3.1.1”、“1.3.1.2”项下方法,取5 g 决明子粉末,精密称定,提取3 次,分别考察液料比(5∶1、10∶1、15∶1、20∶1、25∶1 mL/g)、提取溶剂(40%、50%、60%、70%和80%的乙醇与乙酸乙酯以2∶1 比例混合作为提取溶剂)及提取时间(15、30、45、60、75 min)对决明子总蒽醌提取率或每克决明子提取物相当药材量、5 个指标成分(橙黄决明素、大黄酸、大黄素、大黄酚、大黄素甲醚)的峰面积归一化值[Xi%=(Ai/An)×100%,Ai为同一提取条件下同一样品的5 个指标成分的峰面积和,An为同一提取条件下同一样品的共有峰峰面积和]及对照指纹图谱相似度(“1.3.1.3”项下参照溶液所得指纹图谱为参照图谱S1,以其与供试品溶液指纹图谱生成的对照指纹图谱相似度为准,下同)的影响,并确定综合评价(R)结果。
综合评价(R)=决明子总蒽醌提取率(或每克决明子提取物相当药材量)×0.2+5 个指标成分峰面积归一化值×0.5+对照指纹图谱相似度×0.3 式中,0.2、0.5 和0.3 为权重系数,反映各因素对综合评价的贡献。
1.3.4 Box-Benhnken 响应面优化提取工艺 根据Box-Behnken 试验设计原理,以提取率、峰面积归一化值、对照指纹图谱相似度及综合评价结果为指标,在单因素试验的基础上,选取液料比(A)、乙醇浓度(B)和提取时间(C)3 个影响因素,并利用De‑sign-Expert 8.0 软件设计三因素三水平试验[12],因素与水平表见表1。
表1 响应面试验因素与水平表Tab 1 Factors and levels in Box-Behnken design
2 结果
2.1 指纹图谱的建立及共有峰的确认
按“1.3.1.2”项方法制备决明子总蒽醌供试品溶液,按“1.3.2.1”项下色谱条件进样,记录色谱图,导入国家药典委员会研制的《中药色谱指纹图谱相似度评价系统》(2012.130723 版本)中,建立指纹图谱。以“1.3.1.3”项下决明子总蒽醌参照溶液所得指纹图谱为参照图谱S1,经多点校正,Mark 峰匹配共确认了16 个共有峰;利用决明子总蒽醌供试品中待鉴别组分色谱峰与橙黄决明素等5 种对照品色谱峰保留时间的一致性,确认了决明子蒽醌类提取物的5 个标志物峰[13],分别为橙黄决明素、大黄酸、大黄素、大黄酚、大黄素甲醚,见图1。

图1 决明子供试品溶液(A)与混合对照品溶液(B)的HPLC 图Fig 1 HPLC chromatogram of Cassia seeds test solution(A)and mixed reference solution(B)
2.2 单因素对决明子总蒽醌提取效率的影响
2.2.1 液料比对综合评价(R)的影响 根据预实验结果,确定提取溶剂为60%乙醇-乙酸乙酯(2∶1)混合溶液,提取时间90 m in,考察液料比5∶1、10∶1、15∶1、20∶1 和25∶1(mL/g)时对提取率(%)、峰面积归一化值(%)及对照指纹图谱相似度的影响,并计算综合评价(R)结果。见表2、图2。
由表2、图2 可知,液料比15∶1(mL/g)时,提取率(%)最高、峰面积归一化值(%)最大;各组对照指纹图谱相似度在0.950~1.000,说明各组共有峰数量保持一致,各共有峰峰面积受液料比影响未出现过大或过小波动;决明子总蒽醌提取的综合评价随着液料比(mL/g)的提升而逐渐提高,在液料比15∶1(mL/g)时总蒽醌综合评价得分达到最高0.514。之后随着液料比的增加,总蒽醌综合评价结果逐渐下降。因此,最佳提取液料比为15∶1(mL/g)。

表2 液料比对综合评价的影响Tab 2 Effect of liquid-to-material ratio on comprehensive evaluation

图2 液料比对决明子HPLC 指纹图谱(A)及综合评价(B)的影响Fig 2 Effect of liquid-to-material ratio on HPLC fingerprint spectrum(A)and comprehensive evaluation(B)of Cassia seeds
2.2.2 提取溶剂对综合评价(R)的影响 确定液料比为15∶1(mL/g),提取时间为90 min,分别考察提取溶液为40%、50%、60%、70%和80%的乙醇与乙酸乙酯以2∶1 比例混合作为提取溶剂对每克决明子提取物相当决明子生药含量(简称相当生药含量)(%)、峰面积归一化值(%)及对照指纹图谱相似度的影响,并计算综合评价(R)。见表3、图3。

表3 提取溶剂对综合评价的影响Tab 3 Effect of extraction solvent on comprehensive evaluation
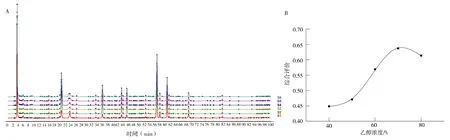
图3 提取溶剂对决明子HPLC 指纹图谱(A)及综合评价(B)的影响Fig 3 Effect of extraction solvent on HPLC fingerprint spectrum(A)and comprehensive evaluation(B)of Cassia seeds
在本次提取溶剂的考察中,乙醇浓度从40%~80%,变化范围较大。乙醇浓度低时,蒽醌苷类成分提取率会增大,而游离蒽醌提取率会降低,同时提取出的水溶性杂质会显著增大;乙醇浓度高时,蒽醌苷类成分提取率会降低,而游离蒽醌提取率会增大。因此,在本次提取溶剂的考察中,并不意味提取物的提取率越高,提取溶剂就越好,因为该提取物的纯度未必较高。因此,采用每克决明子提取物含生药量作为指标,通过极差化法对数据进行处理,使数据落入[0,1]之间[14]。同时,应考虑峰面积归一化值(%)及对照指纹图谱相似度等。
由表3、图3 可知,70%乙醇时,每克决明子提取物相当决明子生药含量最高,但峰面积归一化值并非最高。对照指纹图谱相似度在0.985~0.999,说明各组共有峰数量保持一致,各共有峰峰面积受提取溶剂影响未出现过大或过小波动。计算综合得分,当乙醇浓度为70%时,总蒽醌提取的综合评价得分最高。
2.2.3 提取时间对综合评价(R)的影响 确定液料比为15∶1(mL/g),提取溶剂为70%乙醇-乙酸乙酯(2∶1),分别考察提取时间15、30、45、60、75 min 对提取率(%)、峰面积归一化值(%)及对照指纹图谱相似度的影响,并计算综合评价(R)结果。见表4、图4。
在有关液料比、提取溶剂的考察中,提取时间参照了《中国药典》2020 年版中决明子含量测定项下的提取时间,设置为90 min。该提取时间的设置虽不影响液料比、提取溶剂的考察,但研究发现,在提取3 次的基础上,90 min 设置时间过长。为此,设置提取时间的考察点为15、30、45、60 和75 min。
由表4、图4 可知,不同提取时间点决明子总蒽醌提取率及峰面积归一化值较为接近,其RSD 分别为2.1%和2.2%,对照指纹图谱相似度均大于0.99。综合评价在45 min 达到最高值,考虑到在30 和45min 时,总蒽醌综合评价极为相近,且在后期响应面优化时,45 min 将作为考察时间点三水平上限时间点,因此选择提取时间为30 min。

图4 提取时间对决明子HPLC 指纹图谱(A)及综合评价(B)的影响Fig 4 Effect of extraction time on HPLC fingerprint spectrum(A)and comprehensive evaluation(B)of Cassia seeds
表4 提取时间对综合评价的影响Tab 4 Effect of extraction time on comprehensive evaluation
2.3 响应面优化提取工艺
2.3.1 响应面试验结果及方差分析 根据表1 响应面试验因素与水平表,通过Design-Expert 8.0 软件进行二次响应面回归分析,试验设计及结果见表5,得到多元二次响应面回归模型。
建立决明子响应面17 组试验HPLC 指纹图谱,以“1.3.1.3”项下参照溶液所得指纹图谱为参照图谱S1,利用《中药色谱指纹图谱相似度评价系统》软件对5 个指标成分的峰面积归一化值及对照指纹图谱相似度进行分析计算,结合决明子总蒽醌提取率,确认其综合评价(R)。由表5、图5 可知,不同因素、水平组合条件下决明子总蒽醌提取率的变化范围为20.956%~24.028%,峰面积归一化值的变化范围为32.643%~36.656%,说明提取工艺对决明子总蒽醌提取率及峰面积归一化值影响较大;对照指纹图谱相似度均大于0.99,说明不同提取工艺下各样品与参照溶液在共有峰数量和峰面积总体评价上较为相似。回归模型方差分析结果见表6。

表5 响应面试验设计及结果Tab 5 Response surface test design and results

表6 回归模型方差分析Tab 6 Regression model of analysis of variance

图5 响应面17 组决明子HPLC 指纹图谱Fig 5 Response surface analysis of 17 HPLC fingerprints of Cassia seeds
决明子总蒽醌提取综合评价的二次回归方程为:

由表6 可知,乙醇浓度(B)对总蒽醌的提取呈极显著影响(P<0.01)。模型P<0.05,说明该回归模型是可信的;失拟项P=0.1187>0.05,无显著性差异,说明该模型与实验数据拟合程度较高,存在的误差小,可以用该模型来分析和预测决明子总蒽醌提取过程中各影响因素和响应值之间的关系。F可反映各因素对试验的影响大小,F值越大,表明该因素对试验结果影响越显著[15],由F值可以发现,在所设定的三个因素中,每种因素对决明子总蒽醌提取效率的影响程度为乙醇浓度(F=23.27)>提取时间(F=0.59)>液料比(F=0.26)[16]。模型的残差正态概率分析结果见图6。由图6 可知,模型的残差正态概率基本分布在同一直线上,R2=0.9685,说明决明子总蒽醌综合评价的实际测得值与模型预测值相差较小。

图6 残差的正态概率分布图Fig 6 Normal plot of residuals
2.3.2 验证试验 由Design-Expert 8.0 软件绘制所3D Surface,可以直观地分析各影响因素之间对响应值的影响大小,响应面坡度越陡峭,说明该因素交互作用越显著[17],见图7。以综合评价为指标对决明子总蒽醌的提取工艺进行优化,得到的最佳提取工艺为液料比(A)=20∶1(mL/g),提取溶剂(B)=60% 乙醇-乙酸乙酯(2∶1)混合溶液,提取时间(C)=15.12 min。对优化条件进行5 组验证试验,决明子总蒽醌提取的综合评价得分为0.528(RSD=0.45%),模型预测得分为0.531,与预测得分的相对误差为0.58%,因此该回归模型对决明子总蒽醌提取综合评价的预测准确可靠。

图7 各因素交互作用对决明子总蒽醌提取效果的影响Fig 7 Effect of the interaction of various factors on the extraction effect of total anthraquinone from Cassia seed
3 讨论
将Box-Behnken 响应面法用于中药提取物提取工艺优化的文献较多,评价指标有提取率、有效成分含量等,但未见有将Box-Behnken 响应面法与中药指纹图谱技术结合用于中药提取物提取工艺优化的报道。《中国药典》2020 年版一部中决明子的含量测定项下,以甲醇为提取溶剂,以无水乙醇-乙酸乙酯为大黄酚、橙黄决明素2 种蒽醌化合物含量测定样品的定容溶剂。为此,从提取溶剂价格、毒性、提取率等各方面综合衡量,本试验选择以不同浓度的乙醇与乙酸乙酯(2∶1)混合溶液作为决明子提取物的提取溶剂,采用Box-Behnken 响应面法结合中药指纹图谱技术优化决明子总蒽醌的提取工艺。
Box-Behnken 响应面法可以避免传统整理统计数据存在的弊端,并准确找到最佳的反应条件及灵敏考察各因素之间的相互作用[18]。中药指纹图谱是能够表示中药化学特性共有峰的图谱。通过建立决明子指纹图谱,经Mark 峰匹配,确认了其共有峰,说明在条件优化过程中不会导致某一个成分的缺失,也不会有新的化合物生成。对照指纹图谱相似度的计算是以“1.3.1.3”项下按《中国药典》制备参照溶液所得指纹图谱为参照图谱,结合样品溶液指纹图谱生成对照指纹图谱,对照指纹图谱相似度较高且有所波动。对照指纹图谱相似度波动说明提取条件主要影响共有峰的峰面积,即提取物中各成分的含量;对照指纹图谱相似度较高说明提取工艺变化对各共有峰的影响具有一定的均衡性,不会因提取工艺变化使某个成分过高或过低,各成分峰面积均围绕参照图谱相应的峰面积波动。
提取率是中药提取物工艺优化中常用的评价指标之一,但提取率的设置要考虑提取物的极性及提取溶剂极性的变化情况。本研究是优化决明子总蒽醌的提取工艺,而蒽醌及蒽醌苷是决明子总蒽醌的主要有效物质[19],二者极性不同。提取溶剂极性大,蒽醌苷等极性大的物质的提取率会增大,而蒽醌提取率会降低;提取溶剂极性小,蒽醌苷等极性大的物质的提取率会降低,而蒽醌提取率会增大。因此,在考察不同浓度乙醇-乙酸乙酯对提取工艺影响时,在保证指纹特征相似性及5 种主要指标成分高含量的基础上,采用每克决明子提取物含生药量替代提取率作为评价指标之一更为合理可靠。
在综合评价(R)的计算中,权重系数0.2、0.5、0.3 分别为提取率、指标成分峰面积归一化值和对照指纹图谱相似度对综合评价的贡献。其中,橙黄决明素、大黄酸、大黄素、大黄酚和大黄素甲醚5 种游离蒽醌的峰面积归一化值来自同一提取条件下同一样品的橙黄决明素等5 种游离蒽醌的峰面积和与该样品中16 个共有峰峰面积和的比值,这些数据来自该样品的指纹图谱,是对该样品指纹图谱数据处理的结果,能够综合直观反映提取工艺对5 种指标成分及16 个共有峰的影响,因此设置权重为0.5。指纹图谱作为一种综合的、可量化的质量评价方法,其可全面获得中药化学成分群的整体特征信息。本研究是以《中国药典》制备参照溶液所得指纹图谱为参照图谱,结合样品溶液指纹图谱生成对照指纹图谱,并计算其相似度,考察提取条件变化对对照指纹图谱相似度的影响,因此在峰面积归一化值权重设置的基础上,设置对照指纹图谱相似度的权重为0.3。
本研究对决明子总蒽醌成分的提取工艺进行了优化,可为决明子药效成分的深入研究与应用提供参考与借鉴。
作者贡献度说明:
本文设计思路、提供指导意见及论文审校由邹纯才及鄢海燕教授提供;具体实验过程、数据分析及论文撰写由王恒完成。
