1例Schaaf-Yang综合征与4例Prader-Willi综合征临床特征比较
2022-04-18张晓丽徐发林王慧娟尚利宏李书津
张晓丽,徐发林,王慧娟,尚利宏,李书津
(郑州大学第三附属医院新生儿病区,河南 郑州 450051)
Prader-Willi综合征(Prader-Willi syndrome,PWS)最早由Prader和Willi等[1]于1956年报道,是一种累及多器官的非孟德尔遗传性疾病,由15号染色体q11.2-13 PWS/AS区域父源基因表达缺失所致,临床表现复杂多样,新生儿期肌张力低下,喂养困难,之后出现饮食过度,肥胖,智力低下,身材矮小等[2],是印记遗传的典型代表[3]。MAGEL2是PWS区域的5个编码蛋白基因之一,其截断点突变可导致Schaaf-Yang综合征(Schaaf-Yang syndrome,SYS),由Schaaf等[4]于2013年首次报道。截至目前,全世界共报道81例SYS[5],国内有3例报道[2,6-7]。SYS与PWS有显著的表型重叠,但临床表现有差异,易被漏诊及误诊。本研究对2018年7月至2021年5月就诊的5例患儿的临床资料进行分析,并进行文献复习讨论,以提高临床医生对该病的认识。
1 对象与方法
1.1 研究对象收集2018年8月至2021年4月在郑州大学第三附属医院新生儿科因“反应欠佳、呼吸急促”住院的5例患儿为研究对象,参照Holm[8]等提出的PWS临床诊断标准。
1.2 临床资料采用回顾性研究方法,分析患儿一般情况,包括年龄、性别、胎龄、孕期情况、出生史及家族史等;临床表现为特殊面容、喂养困难、性腺发育不良、肌张力低下等;实验室检查包括血常规、生化、尿常规、血氨、乳酸、同型半胱氨酸、甲状腺功能等。
1.3 基因检测监护人签署知情同意书后,分别采集患者及其父母外周血2~4 mL外送金域、智因东方或医院进行基因检测,采用全外显子组测序、多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)、染色体微阵列法进行检测。
2 结果
2.1 一般资料5例患儿中男1例,女4例;就诊年龄20 min至20 d;均在新生儿期因“呼吸急促反应差”入院。患儿监护人平素体健,均无先天性、遗传性、家族性疾病病史。5例患儿中2例羊水Ⅲ°污染,3例胎儿窘迫;5例为剖宫产娩出;1例为36周早产,其余为足月;除SYS外,其余为小于胎龄儿;均有肌张力低下,呼吸急促,吸吮困难;实验室检查无特殊阳性结果。见表1。

表1 5例新生儿基础资料
2.2 基因型5例患儿中1例为15号染色体MAGEL2基因c.2167delG突变;3例父源性15q11-q13区域缺失型,其中1例为缺失Ⅱ型,2例为缺失Ⅰ型;1例母源混合型单亲二倍体。见表2。

表2 5例新生儿临床表现及预后

表3 1例Schaaf-Yang综合征及4例Prader-Willi综合征患儿遗传学检测结果
2.3 临床表现MAGEL2基因点突变患儿发黑,眉较浓,耳位低,腭弓高,小下颌,双手通贯掌,双手小指及食指压于中指及无名指上,双手不能完全伸展,足趾短,双脚⻊母趾不能自然伸直,隐睾,小阴茎;父源性缺失组患儿均存在肌张力低下,皮肤白皙,哭声弱,吸吮困难;母源混合型单亲二倍体患儿肌张力低下,皮肤白皙,哭声弱,吸吮慢可完成,无特殊面容。见图1。
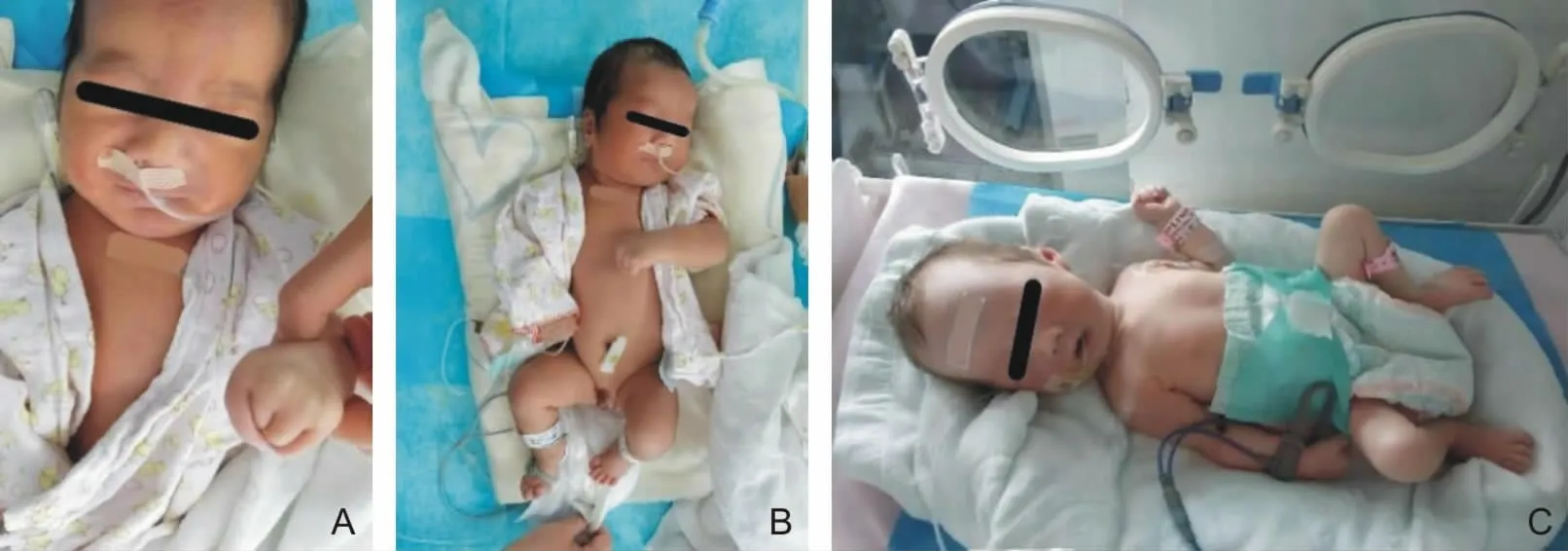
图1 SYS(A、B)和PWS(C)患儿表型特征
3 讨论
有一小部分等位基因只表达其中一个,称为单等位基因表达。对来源于母亲或者父亲的等位基因做一印记使其不表达的现象称为印记基因。来源于母亲的等位基因不表达为母源印记,来源于父亲的等位基因不表达为父源印记[9]。父母的染色体不均等地传递给子代或功能上有所区别,这种因双亲性别不同对后代影响不同的遗传机制称作基因组印记,又称亲代印记或遗传印记。PWS是基因组印记的典型代表,由15q11.2-13区域父源基因表达缺失所致;父源基因可促进胚胎汲取母源能量,最大限度地生长,其调节失控将可能导致胚胎和胎儿的生长发育异常,有研究指出PWS是引起小于胎龄儿的常见微缺失综合征之一[10]。本研究中有3例小于胎龄儿,是否与印记基因相关可做进一步的研究。研究表明人工辅助生殖技术常可使正常妊娠时罕见的印记性疾病增多[9],该4例PWS患儿中最后1例为试管婴儿,人工辅助技术极有可能通过干扰基因印记而影响子代生长发育潜能。
PWS的临床评分诊断系统易受年龄、病程、种族等因素的影响,导致漏诊或延误诊断,因此进行基因诊断并确定其遗传机制很有必要。PWS的发病机制是染色体15q11.2-13区域基因组印记遗传异常所致,长约6 Mb[11]。该区域由3个断裂点分为4个区域,分别为两端的非印记基因区域及PWS、AS区域[12]。该印记表达区域包含一段长序列的父源表达区域(约2 Mb,包括MKRN3、MAGEL2、NDN、C15orf2、snoRNAs和SNRPN-SNURF等基因),一段短序列的母源表达区域(约500 kb,包括UBE3A和ATP10A基因),该区域印记状态的变化受SNRPN基因启动子印记中心微缺失和突变影响[13-14],可由4种不同的缺陷类型所致[12]。(1)父源染色体15q11.2-13片段缺失(西方65%~75%,中国和亚洲人群80%[15]);(2)母源同源二倍体(20% ~30%),母源混合型单亲二倍体已被证实与高龄产妇有关[16],本研究中该患儿母亲年龄为35岁,属于高龄;(3)印记中心微缺失及突变(1% ~3%);(4)染色体平衡易位(<1%)。大多数PWS患儿携带一系列连续基因失活,包括MKRN3(与性早熟相关),黑色素瘤抗原基因(MAGEL2、NDN、NPAP1、SNRPN)以及长链非编码RNA;也有少数病例报道SNORD116 snoRNA簇的微缺失可以观察到PWS的关键表型[17]。PWS的多基因性质使得各基因对其临床表型的贡献变得复杂,不同的分子遗传机制可导致患儿临床症状轻重不同[18]。PWS出生发病率从1/15 000到1/5 000不等,平均病死率3%[19]。本研究中有2例缺失Ⅰ型患儿死亡,其中1例住院期间持续机械通气,吸入氧浓度曾达100%,1周后患儿结果回示PWS,最终家属放弃治疗后死亡。另1例患儿因“呼吸急促20 d,咳嗽3 d”入院,诊断新生儿肺炎,且存在喂养困难,家属签字出院后随访患儿死亡。另1例缺失Ⅱ型患儿现生后2岁,可走路,但语言发育相对落后。目前对于缺失Ⅰ型和Ⅱ型患儿临床表现差异尚未形成统一认识,仍需大样本临床资料进一步明确表型与基因型的关联。有研究报道母源混合型单亲二倍体型患儿面部畸形,睡眠障碍,拼图困难,但语言功能、生长迟缓以及行为异常、智力障碍相对而言较轻[18],精神疾病及自闭症谱系疾病更为常见,可能与UBE3A基因表达增加有关[20],本研究中母源混合型单亲二倍体患儿住院期间喂养可,无呼吸支持,与文献报道[20]一致,后期可进一步随访预后。
SYS综合征是一种极其罕见的遗传性疾病,具有多种肌肉骨骼和神经发育表型,占精神发育迟缓的0.9%。肌张力低下、关节挛缩、精神发育迟缓、面容特殊是对SYS有提示的临床特征。它是一种由位于Prader-Willi关键区印记基因MAGEL2的父系等位基因截断点突变所引起,最早被叫做PWS样综合征。但SYS不像PWS患儿更多的表现为贪食和过度肥胖,其孤独症的发生率更高[21]。MAGEL2功能缺失是导致PWS和SYS患儿严重肌张力减退和肌肉骨骼异常最可能的原因;它是编码黑色素瘤抗原亚家族样2蛋白的唯一外显子基因,编码1 249个氨基酸,是调节内吞、胞饮、受体循环和细胞表面定位的大型泛素化复合物的一部分。PWS患儿因OCA2单倍基因剂量不足,73%的患儿出现色素减退[22]。OCA2基因编码黑素体跨膜蛋白,但在SYS患儿中该基因不受影响。SYS的婴儿也可表现为呼吸衰竭,有研究表明PWS患儿中仅有12%需要气管插管,而在SYS患儿中有33%需要气管插管[23],其呼吸系统并发症是由多种原因引起的,MAGEL2基因在下丘脑中高度表达,但小鼠模型中膈肌和腹壁肌肉也有适度表达,表明功能丧失可能导致低通气[24]。在男性和女性PWS中,性腺功能减退症是一致的,这通常被认为是下丘脑的原因,高达80%的PWS男性被诊断为性腺功能低下,出生时明显为隐睾症[25];而SYS患儿性腺功能减退相对发病率较低,提示MAGEL2基因缺失是性腺功能减退的部分原因。McCarthy等[5]报道的78例患儿中,42例发生突变,最常见的突变是c.1996dupC,该类患儿关节挛缩、喂养困难、呼吸衰竭、精神发育迟缓发病率更高,其严重程度可能取决于截断突变的具体位置。因此,需大样本临床资料的进一步研究以阐明SYS不同表型表达背后的潜在病理机制。
本例SYS患儿的主要表现为唾液黏稠、呼吸性酸中毒、喂养困难和关节挛缩、双手小指弯曲、双侧阴囊小、隐睾和阴茎短小;全外显子组基因测序发现该患儿15号染色体MAGEL2基因c.2167delG 杂合变异(chr15:23890723),且其父母验证未发现该变异,故患儿该变异为致病性变异。生物学分析显示MAGEL2基因c.2167delG会导致氨基酸移码变异以及提前出现终止密码子(p.Ala723Profs*4)。该突变位点尚未在人类基因突变数据库中报道,在大规模人群测序计划数据中也未出现,为罕见变异,丰富了全球MAGEL2基因突变谱。本例患儿结合临床表现与遗传学分析,确诊为SYS,最终该患儿家属签字出院后因呼吸衰竭死亡。MAGEL2基因属于母系印记基因[26],临床医生可能会错误地认为SYS属于常染色体显性遗传性疾病,但健康的父亲可能携带变异的MAGEL2基因,因为印记选择沉默不表达,却将其传给子代而发病,其再发风险为50%。
综上所述,对于新生儿期存在肌张力低下、喂养困难、生长发育迟缓、生殖器发育不全的患儿,需排除PWS,如合并有关节挛缩等表现的,需考虑SYS的可能,可行MLPA及全外显子基因检测,提高确诊率。PWS基本为散发病例,再发风险低,基于不同的遗传机制可进行遗传咨询,对已生育该病患者的夫妇进行再次生育评估及产前诊断,可为早期干预和遗传咨询提供依据。
