组蛋白去甲基化酶GASC1在肿瘤发生中的作用及其抑制剂研究现状
2022-04-18贾瑞诺许冰怡高社干
贾瑞诺,许冰怡,高社干,2
肿瘤发生是遗传和表观遗传学多重改变的结果[1]。遗传改变包括染色体数目和结构变化,基因扩增、缺失和突变。表观遗传改变包括组蛋白修饰和DNA甲基化状态的改变,以及非编码RNA调控的改变等。遗传和表观遗传改变相互依存致使肿瘤发生发展,如基因改变可引起表观遗传改变导致异常染色质调节。全基因组系统分析显示,组蛋白修饰酶(包括去甲基化酶)在多种类型肿瘤中高频[2-4],组蛋白甲基化和去甲基化之间的不平衡致使肿瘤发生[5-6],其核心是组蛋白酶修饰失调。毋庸置疑,更好地理解基因组和表遗传学改变之间复杂的关系,对于认识肿瘤发生和发现新的预后分子标志物和治疗靶点至关重要。
2000年,Yang等从低分化食管鳞状细胞癌(esophageal Squamous cell cancer,ESCC)细胞系KYSE-150扩增区域9p24克隆出一高频扩增基因:鳞状细胞癌扩增基因1(gene amplified in squamous cell carcinoma 1,GASC1),又称KDM4C[2]。随后研究发现GASC1在多种肿瘤中扩增和过表达,并且其表达上调与患者不良预后相关[3-9]。最近,GASC1蛋白已被确定为JMJD2(jumonji domain containing 2)家族成员之一。JMJD2为一组新发现具有转录调节功能的组蛋白赖氨酸去甲基化酶[10-13],而后者在调节基因表达和染色质构架中起重要作用,并由此影响个体生长发育、DNA修复,干细胞生物学行为及肿瘤发生。尤其是组蛋白去甲基化酶,如GASC1,不仅在癌变过程中发挥重要作用,其抑制剂也显示出较强的抗肿瘤能力,在肿瘤靶向治疗方面极富前景[14-18]。
本文将从以下内容综述:组蛋白去甲基化酶GASC1在多种肿瘤中的基因改变;GASC1通过介导组蛋白修饰表观遗传学改变促进肿瘤发生发展的潜在机制;GASC1抑制剂的研发鉴定,以及其在抗肿瘤治疗中的优势及不足。
1 GASC1:食管癌9p24基因扩增区域
基因扩增是人类癌基因激活的重要机制之一,导致其RNA及蛋白过表达[19- 20]。Yang等[2]最初应用比较基因组杂交法分析人食管癌细胞系,发现9p24存在扩展区域。经过检测29个食管癌细胞株,5个KYSE150细胞株(17.2%)被发现存在GASC1扩增。KYSE150是从一位49岁患者的低分化进展期食管鳞状细胞癌培养建立的食管癌细胞系,其在9p23-24区域表现高水平的扩增[2,22]。由于基因扩增区域经常是癌基因或某些肿瘤相关基因的“安乐窝”,并且9p23-24扩增已报道在多种肿瘤中发生,研究者应用荧光原位杂交(fluorescence in situ hybridization,FISH)和DNA印迹(southern blot)分析9p23-24扩增区域,利用Northern印迹法检测该扩增区的靶基因或转录子,表达序列标签克隆(expressed sequence tag,EST)R24542被发现在食管癌细胞系过表达,并在9p23-24区域扩增。应用R24542克隆作为探针在两个不同的cDNA文库筛选,新基因GASC1被成功克隆[2]。
2 GASC1在多种肿瘤扩增和过表达
GASC1在多种肿瘤扩增和高表达,包括淋巴瘤、髓母细胞瘤、肺癌、前列腺癌和乳腺癌等[3-9]。Yang等对一系列乳腺癌细胞系和原发乳腺癌样本进行了全基因组分析,在50例乳腺癌细胞系中发现7例GASC1扩增,包括HCC1954、Colo824、SUM149、 HCC70、HCC38、 HCC2157、MDA-MB-436[3-4,11,23],大约15%原发乳腺癌组织发现该基因扩增[3,11]。7例GASC1扩增细胞系均为侵袭性强、预后极差的基底型乳腺癌亚型[23]。另一研究显示,116例基底型乳腺癌组织相比于83例非基底型乳腺癌组织,GASC1转录表达水平明显增高[3,24]。在35%~45%原发性纵隔B细胞淋巴瘤(primary mediastinal B cell lymphoma,PMBL)及33%的霍奇金淋巴瘤(Hodgkin lymphoma,HL)中也检测到GASC1扩增和/或高表达[25],7.3%髓母细胞瘤发现GASC1扩增[4]。Italiano等利用细胞遗传学、FISH及CGH分析了1例转移性肺肉瘤样癌,在原发灶及两个不同部位的转移灶同时发现GASC1的扩增,表明GASC1是该病例肿瘤发生机制的重要组成部分[9]。
为进一步证实GASC1在不同肿瘤中的扩增情况,Yang等调查CGH阵列数据库,收集了多种癌症类型3 131个拷贝数图谱[26]。在3 131个肿瘤样本中,11.5%存在GASC1扩增,其中2.87%表现为GASC1基因局灶性扩增峰值,特别在乳腺癌和肺癌。基于癌症基因组鉴定重要靶点(genomic identification of significant targets in cancer,GISTIC),243例乳腺癌标本有15.64%GASC1扩增,其中4.53%高水平扩增。在774例肺癌组织标本中13.44% GASC1扩增。在其他肿瘤中也频发出现GASC1扩增,如食管鳞状细胞癌20.45%,卵巢癌19.42%,大肠癌19.25%。见表1。另有报道显示,相对于正常前列腺组织,GASC1在前列腺癌组织中显著过表达[10,27]。
3 GASC1的恶性转化潜能
GASC1在多种细胞模型表现具有恶性转化潜能。为证实GASC1可否引起人正常乳腺上皮细胞恶性表型转化,野生型GASC1经慢病毒包装转染正常乳腺上皮细胞MCF10A,结果显示GASC1过表达导致细胞获得恶性转化表型,包括生长因子非依赖性生长和悬浮成球培养阳性[3]。在人工基膜Matrigel三维立体培养形态形成实验中,MCF10A细胞形成具有极性特征、有限生长、空腔样正常类腺泡样结构;而MCF10A-GASC1细胞则多呈异常腺泡样的实性内腔结构[3]。这些结果表明,GASC1过度表达可妨碍上皮细胞正常组织结构,而这种现象是肿瘤形成始动阶段的常见改变。
利用基因敲除方法证实GASC1对于乳腺癌、食管癌、前列腺癌和淋巴瘤等多种肿瘤表型维持起重要作用。在GASC1扩增乳腺癌细胞系HCC1954、Colo824和SUM149,利用GIPZ表达阻滞shRNA慢病毒系统稳定敲除GASC1表达,与MCF10A细胞相比,敲除GASC1可显著抑制乳腺癌细胞增殖及克隆形成[3]。利用相同方法显示敲除GASC1同样抑制食管癌细胞KYS150增殖及克隆形成[10]。在前列腺癌,GASC1与雄激素受体(androgen receptor,AR)相互作用,是AR诱导转录的共激活子,利用shRNA下调GASC1表达可抑制雄激素依赖性前列腺癌细胞增殖[28]。前文提及,淋巴瘤尤其是PMBL a和HL,9p24区域呈高频扩增。为识别这一扩增区域的癌基因,Rui等在PMBL和HL中利用RNA干扰筛查了9p24扩增区域功能性关键基因。他们发现GASC1和JAK2共同维持淋巴瘤细胞增殖和存活[25]。总之,GASC1作为癌基因在多种类型肿瘤发生发展中起重要作用。
4 GASC1催化组蛋白去甲基化
染色质修饰作为表观遗传调控的重要机制,成为近几年的研究热点,组蛋白修饰异常调节与恶性肿瘤发生发展密切相关[1,6]。染色质基本单位是核小体,其由147bpDNA碱基以不断重复的核小体核心:4对组蛋白H2A、H2B、H3和H4为轴心盘绕而成。这些组蛋白呈球形,但有一包含了大量转录后修饰的N末端。组蛋白末端修饰包括甲基化、乙酰化、磷酸化、泛素化及异构化,从而结合蛋白标志,被称为组蛋白密码[29]。组蛋白赖氨酸甲基化受组蛋白甲基转移酶和去甲基化酶动态活化所调控,其作为主要染色质调控机制影响着根本的核转录过程,并在转录后调控中起核心作用[15,29]。不同赖氨酸残基的甲基化、同一赖氨酸残基的甲基化程度以及在特殊基因位点的组蛋白甲基化,将导致不同的转录和生理后果。研究显示在组蛋白H3的5个位点上(K4、K9、K27、K36、K79)发生赖氨酸甲基化对基因转录有明显影响[15-16,30]。一般来说,H3K4、H3K36和H3K79甲基化与转录激活相关,而H3K9和H3K27的甲基化(H3K9me3/me2)(H3K27me3/me2)与转录抑制相关[30]。
GASC1于2000年最初被克隆时,其被预测可能是一种介导染色质转录调控的核蛋白,但并不清楚其作用机理,包括在正常细胞和肿瘤细胞中如何调控转录。2004年,Katoh等确定GASC1属于Jumonji亚家族JMJD2成员之一[31]。该研究定义GASC1为JMJD2C,含有一个Jumonji(Jmj)C结构域,一个JmjN结构域,两个植物同源结构域(plant homeo domain,PHD)型锌指和两个Tudor结构域。2006年,随着对JmjC结构域蛋白的鉴定,人们在理解染色质调节机制方面出现新的突破,包括对GASC1的认识,明确其为一种新的组蛋白去甲基化酶[10,13,32]。2007年,GASC1被命名为KDM4C(lysine-specific demethylase 4C)[33]。JmjC家族由30个同源子组成,18个已明确具有组蛋白去甲基化酶活性,并进一步分为7个亚家族(kdm2-8)[15]。人类KDM4亚家族有6个成员(KDM4A-F),其中2个成员KDM4E/F,可能是假基因[31]。KDM4A、B和C(GASC1)蛋白,有超过50%序列同源性,包含JmjN、JmjC、PHD和Tudor结构域,但KDM4A缺乏羧基端PHD和Tudor结构域。见图1。
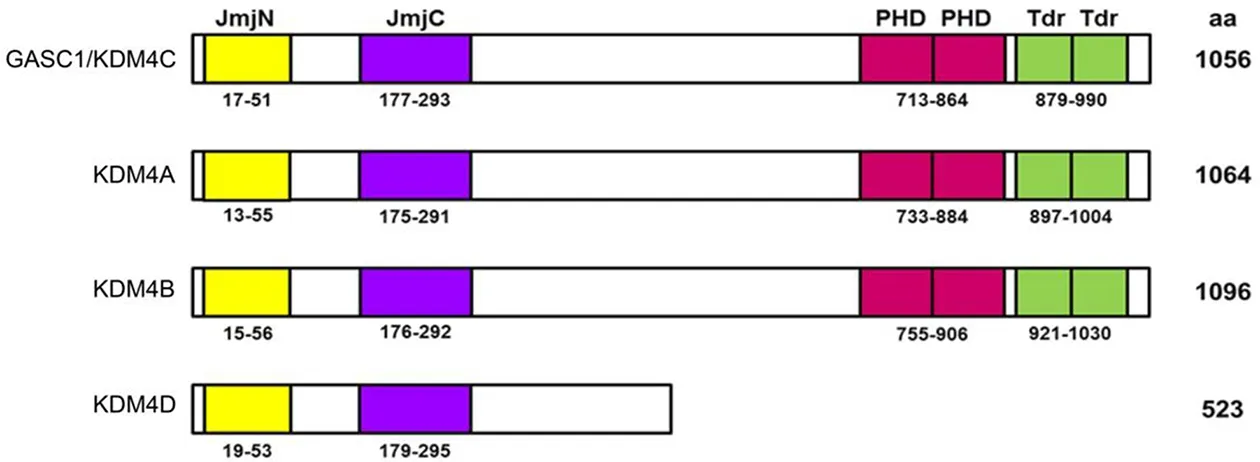
图1 GASC1及其同源子KDM4A、B和D结构域的位置和长度(基于美国国家生物技术信息中心NCBI数据)
GASC1N-末端的Jumonji域是组蛋白去甲基化酶的催化核心[10,13,32]。Jumonji,在日文中名“十字”,人们将在小鼠体内可引起神经板消融变性形成酷似十字状的这类转录因子命名为Jumonji[34]。根据在蛋白中的相对位置,JmjN和JmjC被认为是Jumonji蛋白中的保守序列[35-37]。JmjC结构域是GASC1作为组蛋白去甲基化酶的催化域,JmjN提供和JmjC形成广泛相互作用的完整结构[12,14,32]。JmjC及JmjN是所有含JmjC结构域蛋白(包括GASC1)实现去甲基化的基本功能单位[12]。GASC1 Jumonji结构域介导的去甲基反应是双加氧反应,依赖Fe(Ⅱ)和a-酮戊二酸(a-ketoglutarate,a-KG)作为共刺激因子参与,见图2。体外及细胞学研究显示,GASC1Jumonji结构域可催化H3K9me3/me2和H3K36me3/me2去甲基[12-14]。综合结构、生化及细胞研究表明,GASC1可催化H3K9me3/me2去甲基化(H3K9me3/me2减少4~5倍),并有效减少H3K36me3/me2底物[38]。酶底物竞争性试验显示GASC1对H3K9三甲基多肽的结合明显优于二甲基[38]。GASC1催化域(1-347aa)的晶体结构可以在Protein Data Bank (PDB)数据库查获(2XML)。体外生化及细胞学分析显示,GASC1JmiC结构域的H190、E192及H288残基是组成Fe(Ⅱ)结合槽的基本单位,H190和E192突变可明显降低H3K9去甲基化活性[10]。因此,GASC1的Jumonji结构域主要使H3K9me3去甲基化,其C-端PHD和Tudor域可能有助于GASC1有效的核定位和介导GASC1与组蛋白和其他蛋白质结合[39]。然而,亚基PHD和Tudor域在GASC1依赖的染色质调控机制中的确切作用仍不明了。
5 GASC1参与肿瘤生成的分子机制
H3K9和H3K36二甲基和三甲基是调控转录和其他生物进程的重要标志,因此推测GASC1表达异常可导致组蛋白甲基化途径失衡,影响转录过程中核染色质调控、DNA修复、染色体稳定性,这些均为肿瘤发生最相关的分子机制。GASC1通过H3K9去甲基化作用可上调重要癌基因MDM2和myc表达,以及干细胞关键转录因子NANOG[3,25,40-42]。Ishimura等利用小鼠胚胎成纤维细胞(mouse embryonic fibroblasts,MEFs)寻找GASC1调控下游靶基因[40],外源性过表达GASC1在MEF细胞,发现MDM2 mRNA和蛋白水平表达增加,染色质免疫沉淀(chromatin immunoprecipitation,ChIP)分析示GASC1被招募到MDM2基因启动子区P2,致MDM2的H3K9me3/me2去甲基化,但该研究没有检测到随着GASC1过表达,MDM2 P2启动子H3K36me3水平变化。利用siRNA敲除GASC1表达可引起MDM2表达下调。Wissmann等首次确定GASC1作为H3去甲基化酶调节AR功能[28],通过体外和体内GASC1与AR结合试验、组装AR配体使GASC1结合AR启动子,结果导致H3K9me3去甲基化,使雄激素受体依赖的转录上调。相反,GASC1敲除可抑制雄激素H3K9me3去甲基化和转录激活。Rui等研究发现,在淋巴瘤细胞敲除GASC1,可通过直接改变myc基因启动子和内含子1区H3K9me3而抑制myc表达。
肿瘤干细胞(tumor stem cell,TSC)学说认为肿瘤细胞内存在着一群“干性”细胞,负责肿瘤生长、转移和复发[43-44]。多能胚胎干细胞(embryonic stem cells,ESCs)H3K9甲基化状态就是通过转录因子和组蛋白去甲基化酶活性间复杂的相互作用而维持[42,45]。关键性转录因子,包括OCT4(POU5F1)、NANOG和SOX2,形成一个强大的自动调节通路,维持着胚胎干细胞自我更新[46]。GASC1优先表达在未分化ESCC[47]。2007年,LOH等明确在小鼠胚胎干细胞,GASC1作为OCT4的靶基因,并识别了NANOG是GASC1靶基因之一:GASC被招募到NANOG启动子[33],进一步证实GASC1是胚胎干细胞NANOG基因启动子区逆转H3K9me3所必需的。通过Loss功能研究发现,GASC1对于胚胎干细胞维护至关重要。上述内容提示GASC1作为癌基因,可能通过调节肿瘤干细胞表型参与肿瘤发生发展。但去甲基化酶GASC1在肿瘤干细胞中的作用机制尚需进一步研究,诸多问题等待回答。然而,最近众多研究支持该假说:GASC1扩增和产物过表达,可诱导组蛋白甲基化表观遗传学修饰改变,从而影响在癌生成和人类干细胞维持中起关键作用的基因表达。
6 GASC1:极具潜能的靶向治疗
组蛋白去甲基化酶包括GASC1在肿瘤发生过程中,通过表观遗传学修饰发挥关键作用,为研发去甲基化酶抑制剂作为一类新抗癌药物提供了科学依据[16-17,48-49]。以GASC1/JMJD2家族组蛋白去甲基化酶三维结构和催化机制研究为基础,大量JMJD2抑制剂被鉴定和报道,以下主要综述GASC1抑制剂。
GASC1是Fe(Ⅱ)和a-KG依赖性酶,通过氧化甲基化的组蛋白赖氨酸残基,从而实现使其去甲基化(图2)。靶向GASC1激活所必需的辅因子为我们提供了第一个有意义的结果。2006年,Cloos等首次报道了noxalylglycine(NOG)作为a-KG类似物,能够轻微抑制GASC1对H3K9me3去甲基化的活性[10],该研究推测NOG作为a-KG的类似物,代替a-KG与GASC1铁结合残基结合,而抑制GASC1脱甲基活性。基于GASC1 Jumonji域复合体a-KG的晶体结构模型,Hamada等合成一系列GASC1小分子抑制剂,后命名NCDM-32是选择性最强的GASC1抑制剂[19,50],与NOG比较,NCDM-32显示出非常低微摩尔IC50值,高于NOG500倍的抑制效力[50]。利用生化结构和细胞学分析,Chowdhury等发现2-羟基戊二酸(2-Hydroxyglutarate,2HG)可抑制a-KG依赖性加氧酶活性,包括GASC1[51]。Methylstat,一种细胞活性选择性组蛋白去甲基化酶抑制剂,可抑制三甲基赖氨酸去甲基化酶家族成员。这种小分子抑制剂通过一个连接结合含有甲基赖氨酸底物的类似物,即a-KG辅因子模拟物。Methylstat通过抑制GASC1扩增明显抑制了KYSE150食管癌细胞生长,1/2最大生长抑制浓度(GI50)约为5.1 μM。
利用高通量质谱芯片对10万种以上化合物作为GASC1候选抑制剂筛选,该芯片以小氨基酸多肽为底物,筛选与组蛋白H3共反应前15个氨基酸残基,并直接检测底物三甲基Lys9脱甲基形成的二甲基Lys9产物[52]。其中1 126种IC50值小于100 μM化合物被发现,例如含有8-羟基喹啉(8-hydroxyquinolines,8HQs)核心结构的某些化合物,可以表现出强烈抑制GASC1脱甲基酶活性潜能(IC50:2.1 μM)。8HQs通过结合KDM4A活性位点Fe(Ⅱ)而发挥抗KDM4去甲基化活性。通过对640个自然产物文库筛选,Nielsen等利用甲醛脱氢酶方法检测了21个化合物,发现咖啡酸为有效GASC1抑制剂[53]。咖啡酸为抗氧化剂,既往研究表明其通过影响氧化过程抑制肿瘤细胞生长[54]。最近,一种杂环系统文库被用来筛选新GASC1抑制剂,一个4-羟基吡唑支架被认为可能是一种新KDM4C抑制剂[55]。
7 展望与结论
恶性肿瘤既往被视为遗传性疾病,越来越多的证据表明表观遗传学修饰在肿瘤发生的每一个阶段起着至关重要的作用,并对其治疗产生深远影响。表观遗传学和遗传学的共同作用与相互关联在肿瘤发生发展中的重要作用日趋明了。GASC1最初在ESCC基因扩增区域发现并克隆,至今已被确定为在表观遗传组蛋白修饰起关键作用的组蛋白赖氨酸去甲基化酶。GASC1在多种恶性肿瘤和体外转化表型模型中扩增和过表达,但许多重要问题仍未明确,如GASC1依赖的染色质修饰转录调控对于肿瘤发生发展影响的分子机制等。进一步理解GASC1对染色质组成和转录的影响,以及GASC1缺失和过表达对于转录和组蛋白修饰的作用,将是决定GASC1全基因靶点的关键。研究GASC1在不同肿瘤是否靶向作用于不同基因至关重要。同时,GASC1还可对非组蛋白底物(与H3K9具有相似序列)去甲基化[56-58]。最近研究显示,GASC1可作用于染色体框同源子4(chromobox homolog 4,CBX4,又名多梳2蛋白,polycomb 2 protein:Pc2)的K191me2脱甲基。CBX4在细胞周期和生长控制方面发挥重要作用[58]。然而由GASC1介导的组蛋白与非组蛋白去甲基化之间的相互作用未有报道。
基于GASC1在肿瘤发生中的关键作用,GASC1抑制剂有望很快进入临床试验。然而,大部分GASC1抑制剂的分子支架来源于其他结构或机制相关的酶或蛋白,因此有时候其可能抑制其他酶家族。另外,许多抑制剂是辅因子和/或底物模拟物,因此对于GASC1特异性抑制作用有限,或不确定。因此,未来的研究中,致力于发现高选择性、特异性靶向GASC1的抑制剂至关重要。
