基于TAIL-PCR分析外源基因在小鼠精原细胞中的整合位点
2022-04-02冯美莹曾梓凡邝乃诵温淑桦刘文华王瑛华
冯美莹,曾梓凡,邝乃诵,温淑桦,刘文华,王瑛华
(肇庆学院生命科学学院,广东 肇庆 526061)
外源基因在动物细胞中的表达受到很多因素影响,其整合位点会影响细胞的生长状态及受体动物的表达效应,确定外源基因的整合位点在转基因动物的研究中非常重要[1].外源基因的整合是随机的[1-2],发生在细胞周期的不同阶段时,其整合效率和转入纯度都不一样[3].只有发生在染色体分离的DNA合成期,外源基因会均匀地整合到每一个细胞中;外源基因整合发生在细胞周期的其他阶段时,则会发生嵌合[2].因此,在转基因细胞的研究中,确定外源基因的整合位点是对外源基因表型研究和功能探讨的前提条件.
在研究转基因细胞外源基因的整合位点时,最直接和有效的方法是克隆转基因整合位点及其邻近宿主基因组的序列,并将其与宿主细胞的基因组碱基进行比对,进而确定其转入宿主细胞的位置[4-5].然而,在进行PCR 扩增时,常常由于DNA 含量有限而增大检测难度.热不对称交互式PCR(Thermal asymmetric interlaced PCR,TAIL-PCR)技术可以不受DNA含量的限制,结合特异引物和简并引物进行3轮PCR扩增,简单有效快速地回收已知序列的邻近DNA片段,进而分析外源基因在宿主基因组的整合位点[6-7].TAIL-PCR技术被广泛地应用在转基因动植物中实现转基因动植物外源基因的整合位点序列分析[3,6-7].本研究运用TAIL-PCR技术确定外源基因Cas9在小鼠精原细胞中的整合位点,探究外源基因整合位点的特征,为转基因动物纯合体的育种和外源基因定点整合的研究提供依据.
1 材料与方法
1.1 试验材料
试验所用的材料来源如下:细胞总RNA提取试剂盒购于Tiangen公司,实时荧光定量PCR扩增Mix和E.coli-DH5a 感受态购于南京诺唯赞公司,pMD19-T 载体、DNA 聚合酶和凝胶DNA 回收试剂盒等购于Takara公司,细胞培养所用的试剂购于GIBCO公司,其他试剂购于广州鼎国生物技术有限公司.载体测序由广州艾基生物公司完成.
试验所用溶液配方如下:细胞完全培养液中含有DMEM(Dulbecco’ s modified Eagle medium)、体积分数为10%的胎牛血清(FBS,Fetal Bovine Serum)和4 mmol-谷氨酰胺;LB液体培养基中含有1 g酵母提取物、2 g蛋白胨、2 gNaCl和200 mL纯净水(其中添加20 mg氨苄青霉素可配成LA液体培养基,添加1.5 g琼脂粉和20 mg氨苄青霉素可配成LA固体培养基).
1.2 试验方法
1.2.1 动物贴壁细胞培养
将细胞从液氮中取出,37℃水浴融化和离心后,加入完全培养基重悬细胞,并转移至培养皿中,置于5%的CO2培养箱中培养.待细胞密度达到80%~90%时,加入2 mL胰蛋白酶消化,完全培养基终止后,同样是离心,弃上清,加入完全培养基重悬,完成传代.
1.2.2 细胞基因组抽提
待细胞长满后按上述步骤消化处理,磷酸盐缓冲液(Phosphate Bufferd Saline,PBS)洗2遍后留下细胞沉淀.然后,加入蛋白酶试剂和细胞裂解液70℃热处理10 min.接着,加入无水乙醇沉淀基因组,并利用吸附柱收集细胞基因组,漂洗液清洗吸附柱上的离子和蛋白残留后,50 μL的TE缓冲液或dd H2O洗脱吸附柱上的细胞基因组,-20℃冰箱保存备用.
1.2.3 TAIL-PCR扩增
运用表1中的特异性引物和简并引物按照刘耀光的反应条件进行3轮半巢式PCR扩增[8].第一轮PCR反应以小鼠精原细胞基因组DNA为模板,使用6个DNA熔解温度(Melting temperature,Tm)为68℃的嵌套式特异性引物(Specific primer,SP)与4 个低Tm 的简并引物(Arbitrary degenerate primer,AD)[8]随机组合对外源基因3’ 下游和5’ 上游侧翼序列进行PCR扩增.前一轮的PCR产物稀释100倍用于后一轮PCR反应,第二轮和第三轮分别使用特异性引物与简并引物共有序列(AC引物)进行组合,使特异性的产物被选择性扩增.

表1 PCR引物序列
然后,制作1%的琼脂糖凝胶进行电泳,在凝胶成像分析仪分析3轮PCR 的扩增片段,并按照凝胶DNA微量回收试剂盒的操作回收第三轮PCR的特异片段,-20℃冰箱保存备用.
1.2.4 T载体克隆
首先,将回收的目的片段分别按照“pMD19-T Vector 1 μL、目的片段0.3 pmol,加水补充到5 μL后,加入5 μL的Solution I”的步骤将目的片段与pMD19-T载体进行连接,短暂离心后,置于16℃反应30 min.然后,全量加入100 μL 的E.coli-DH5α感受态细胞中,冰上反应30 min 后,42℃热激60 s 且冰上冷却3 min,并加入300 μL的LB培养液,置于37℃摇床(100~150 r/min)震荡培养1 h.接着,取100 μL菌液涂布在含有氨苄青霉素的LA 固体培养皿中,37℃培养12~16 h.最后,在LA 固体培养皿上挑选单克隆菌落接种于300 μLLA 培养液中,在37℃摇床(220 r/min)中扩大培养3~4 h.
1.2.5 菌液PCR检测
以1 μL的菌液为模板,使用M13通用引物和DNA聚合酶对菌液进行PCR鉴定,反应体系如下(10 μL):5 μL 2×Phanta Max Buffer、0.2 μL dNTP Mix、1 μL 菌液、0.4 μL M13 上游引物、0.4 μL M13 下游引物、0.2 μL Phanta Max Super-Fidelity DNA Polymerase(1 U/μl)和2.8 μL dd H2O.短暂离心后按照“95℃预变性3 min、95℃变性15 s、60℃退火15 s、72℃延伸30 s和72℃总延伸7 min”的程序进行,其中变性到延伸的步骤循环35次.随后,进行琼脂糖凝胶电泳检测,并挑选正确的菌落进行测序.
1.2.6 生物信息学分析
将测序序列拼接后分别在NCBI 用Blastn(https://blast.ncbi.nlm.nih.gov/Blast.cgi PROGRAM= blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome)和 在Ensembl 用BLAST/BLAT search(http://asia.ensembl.org/Mouse_sapiens/Tools/Blast)与小鼠基因组进行同源性序列比对,从而分析外源基因的整合位点.
2 结果与分析
2.1 小鼠精原细胞的培养
稳定表达Cas9的小鼠精原细胞刚复苏时呈圆形、悬浮的状态,经过3~4 h培养后开始贴壁生长,胞体向外伸出呈梭形或不规则形状(如图1),12 h后基本完成贴壁生长,细胞密度逐渐增大.由图1也可以看出,稳定表达Cas9的小鼠精原细胞不仅生长速度快,而且折光性好,细胞状态良好.
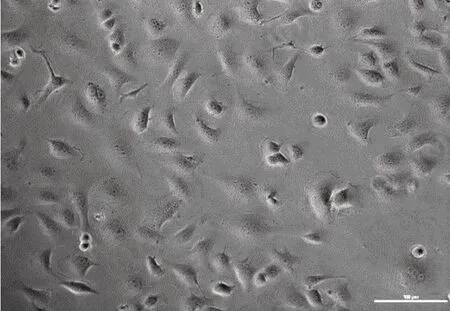
图1 小鼠精原细胞贴壁情况(标尺=100 μm)
2.2 Cas9基因表达检测
为了研究Cas9基因的转入情况,我们以稳定表达Cas9的小鼠精原细胞基因组作为阳性对照,未转入Cas9的小鼠精原细胞基因组作为阴性对照,通过PCR扩增对结果进行分析.如图2A 所示两组细胞的基因组提取效果良好,经过琼脂糖凝胶电泳检测发现细胞基因组条带.通过对目的基因片段进行PCR 扩增后发现在500~750 bp 处,阳性对照中出现目的条带,符合Cas9 引物设计时的长度639 bp 范围,阴性对照中则没有(图2B).由此可见,PCR 扩增所得的基因为目的基因,Cas9基因成功转入到小鼠精原细胞的基因组中.

图2 Cas9基因PCR扩增结果
2.3 阳性对照组的小鼠精原细胞基因组扩增
为了寻找Cas9基因转入到小鼠染色体中的位置,将特异性引物与简并引物结合起来利用TAIL-PCR技术对小鼠基因组进行扩增.设计特异性上游引物检测时,结合简并引物经过3轮PCR后在第二个和第四个产物(图3C的2和4)中出现特异性条带(图3A、B 和C);设计特异性下游引物检测时,结合简并引物经过3 轮PCR 扩增后4 个孔中均出现特异性条带(图3D、E和F).这些都有可能是Cas9转入到小鼠基因组的位置,将其回收后进行下一步的分析.图3中A、B和C为特异性上游第1、2和3轮的PCR扩增,图3中D、E和F为特异性下游第1、2和3轮的PCR扩增.其中,M表示marker,图A和图D的1、2、3和4分别是SP1和SPA1与4个简并引物的扩增片段,图B和图E的1、2、3和4分别是SP2和SPA2与AC引物的扩增片段,图C和图F的1、2、3和4分别是SP3和SPA3与AC引物的扩增片段.

图3 TAIL-PCR扩增结果
2.4 Cas9基因的染色体定位
为了进一步确认Cas9基因转入到小鼠基因组的位置,我们将上述结果回收的片段连入T 载体中,通过PCR扩增筛选出阳性的单克隆菌落(图4 A),并将其与小鼠基因组序列进行比对,从而分析其整合位点.结果发现,6个回收片段中,有2个(图3F的1和2)是属于小鼠基因组序列,其余的为载体序列(结果未展示).经过比对分析,这2个片段均定位在小鼠基因组的12号染色体中(图4B 和C),这表明Cas9基因整合到小鼠基因组的12 号染色体上.图4A 为特异性下游第二个片段的菌液PCR检测结果,图4B为特异性下游第二个片段的与小鼠基因组的比对结果,图4C 为特异性下游第二个片段的定位到小鼠染色体位置.
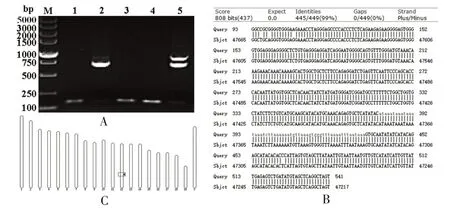
图4 Cas9基因的整合位点分析
3 讨论与结论
在目前转基因动植物整合位点的研究中,用于获取转入位点两端未知基因组序列的方法众多[2-4,9],接头PCR在酶解基因组中接上已知序列,可提高扩增效率,但特异性片段少,且操作步骤繁杂[10-11];质粒拯救需要完整地复制起点和抗性筛选基因序列,且产物过长,基因组自连接和克隆转化的操作效率低,试验难度大[12];这2种方法并不适合用于探讨外源基因在小鼠精原细胞中的整合位置.iPCR法较方便和费用低,常被用于载体构建[13-15]、启动子扩增[16]、外源基因整合位点分析[17-18],但是iPCR需要对基因组进行酶切和环化,要求宿主基因组具有较高的浓度和纯度,而且操作和结果不确定性高,时常出现没有结果的情况.早期运用该方法研究外源基因转入小鼠精原细胞的染色体位置时,常出现没有结果或者基因组丢失的现象(结果未展示).经过多次尝试无果后,本研究采用特异性高和操作简单的TAIL-PCR法,利用特异性引物和简并引物进行扩增,使用少量的宿主基因组也可以有效、快捷的找出外源基因的整合位置[6-7,19].在研究细胞永生化载体元件插入位置时,黄惠等扩增出永生化细胞基因组的侧翼序列,将其与宿主基因组进行比对分析发现使细胞永生化的hTERT基因随机整合于2号、4号、6号、12号、13号、21号染色体中[20].本研究利用TAIL-PCR同样在有限的小鼠精原细胞基因组中克隆出外源基因整合到基因组的旁侧序列,并发现其整合到第12号染色体中,为以后转基因动物纯合体的育种研究提供依据.
此外,TAIL-PCR法常被广泛应用于克隆启动子序列和基因全长[21-25]、扩增基因组侧翼序列[26-29]、分析转基因植物和动物的拷贝数和纯合度[3,5,30-31]等领域中,并在转基因细胞和动物中用于分析外源基因的整合位点及其对宿主生物功能的影响.在分析外源基因整合位点的纯合性中,孔庆然等采用该技术成功在绿色荧光蛋白转基因猪中克隆出TgInS1(1 440 bp)、TgInS2(1 263 bp)和TgInS3(1 861 bp)这3 个整合位点,从而确定纯合的转基因猪[30],这与本研究运用该技术在动物转基因细胞中检测外源基因转入位置有着异曲同工之效,也对于以后研究转基因动物的纯合性有着重要的指导作用.同样,TAIL-PCR还能有效应用于外源基因整合在转基因植物中的研究.运用TAIL-PCR检测发现,突变体黄瓜扩增的T-DNA侧翼序列插入到参与气孔开放过程的Bhp基因的第10个外显子中[32],抗虫转基因玉米的外源基因整合到玉米基因组第5号染色体中[33],拟南芥突变体基因组的突变发生在影响维管发育的基因At1g 52 910第2外显子中[34].由此可见,在转基因植物中同样可以与本文一样利用TAIL-PCR 在序列复杂的转基因事件中鉴定出基因插入位置,而且利用TAILPCR技术能够很好地分离已知基因的侧翼序列,对于研究外源基因在转基因动植物中的整合位点确认及相关生物学功能具有重要的意义.
