基于深度学习的化学结构识别研究
2022-03-30蚁佳才张小琛刘丹国防科技大学计算机学院长沙410073
蚁佳才,张小琛,刘丹(国防科技大学计算机学院,长沙 410073)
科研工作者通常将经过大量实验收集的化学知识转化为文献中的图文描述,这些海量的知识是化合物重新发现及药物发现研究的巨大财富。文献中的化合物结构是药物发现的核心,而这些结构信息在大多数情况下是以图像的形式呈现,这些有价值的信息对于机器来说是非结构化且不可读的。因此,自动化地将化学结构由图像转换成人类或机器可读的格式,对于从大量文献中挖掘知识是非常重要的,该过程被称为化学结构识别(chemical structure recognition,CSR)。
对于机器可读的分子结构,目前采用广泛的包括国际通用技术和统一标准的线性码简化分子线性输入系统(simplified molecular input line entry system,SMILES)以及分子的三维表示,如图、矩阵或连接表等。SMILES可以减少存储空间,提升检索效率,并且可以很方便地编码分子结构、性质信息以及转换成其他三维表示,因此被广泛应用于各种与化合物分子相关的研究中。在CSR中,其目标是将化学结构图像转换为相应的SMILES字符串,即图像到文本的转换。图1展示了阿司匹林(aspirin)的化学结构以及相应的SMILES,从图中可以直观地看出,除了识别出图像中相应的元素以外,还需要根据相应重构算法将这些元素的符号组合成有效的SMILES。

图1 化学结构识别示意图Fig 1 Diagram of chemical structure recognition
本文采用深度学习的方法,基于编码器-解码器架构,融合注意力机制和分子指纹先验知识,构建了一个深度学习模型——基于指纹预训练的化学结构识别(chemical structure recognition based on fingerprint pre-train,CSRFP),现报道如下。
1 相关研究工作
1.1 基于规则的方法
20世纪90年代初,第一个完整的化学结构识别系统Kekulé发布了。Kekulé是一个基于规则的工具,通过将单一化学结构图进行分割、向量化、图像分块、图像腐蚀、图像膨胀以及光学字符识别等一系列步骤,最终将化学结构图转换为新的分子图表示。随后,越来越多的研究人员开始关注这一领域,研发出了各种开源软件。Rajan等总结了该领域的研究进展,并比较了3个基于规则的开源软件OSRA、MolVec和Imago,为未来的深入研究提供了参考。传统的分子图像识别方法在很大程度上依赖于专家知识,并且需融合新的待检测图像特征,费时费力。另外,这些工具通常步骤繁琐,导致计算时间较长。具体来说,大多数基于规则的方法往往包括以下步骤:① 扫描,即扫描包含化学结构图的页面区域,并将其从页面中分割出来;② 矢量化,即将位图转换为矢量图;③ 搜索虚线和楔形线,即使用专门的算法来识别立体化学信息,如虚线键和楔形键;④ 光学字符识别,即使用光学字符识别(optical character recognition,OCR)算法识别原子和常规键。并结合一些工具使用规则来纠正OCR引擎不能正确识别的原子或原子组;⑤ 图编译,即将原子视为节点,将化学键视为边,并通过图重构算法将其编译成分子图;⑥ 后处理,例如将一些超原子转化为原来的原子序列。
1.2 基于深度学习的方法
近年来,深度学习在计算机视觉领域取得了较大突破,如图像分类、目标检测、语义分割以及图像字幕等。CSR可以视为特殊的图像字幕任务,与自然语言文本生成不同,CSR的条件更为严苛,例如其生成的元素字符往往出现多个重复元素,并且上下文信息联系更加紧密,且元素字符具有唯一性,而自然语言往往可以使用多种同义词进行替换。基于深度学习的方法的优点在于它们不需要制订复杂的规则,模型可以自动拟合学习高维特征空间,并且具有更好的泛化能力等。
目前基于深度学习的方法主要包括MSEDUDL、Chemgrapher以及DECIMER。其中,MSE-DUDL主要研究如何将文献中的化学结构进行分割并识别成机器可读的形式。其使用一个类似U-Net的分割网络从文献中分割出化学结构图,并使用卷积神经网络(convolutional neural network,CNN)结合循环神经网络(recurrent neural network,RNN)进行预测。作为两阶段模型的代表,Chemgrapher则使用一个分割网络来识别每个像素的类型,包括原子、键和电荷,并使用一个预测网络预测每个被识别的像素的类型,包括原子、键和电荷类型。两阶段模型可以有效地减少分子图像识别的噪声,但它们可能会受到与优化有关的问题的影响,如误差传播。DECIMER使用一个预训练好的Inception V3模型作为特征提取器,提取图像特征后进行RNN解码。而这种方式下,特征提取器和RNN没有进行联动学习,模型不能学习到有价值的化学图像表征,导致模型的识别率较低。目前,基于深度学习的方法有的对计算资源要求极高,有的标记数据集难以获得,而有的识别精度则较低。
融合先验知识的方法在许多任务中都展现出了非常好的效果。在本研究中,我们首先构建了一个预训练模型来学习化学图像的指纹表示,并将该训练好的模型作为下游任务的编码器进行微调。分子指纹是一种人工编码,其包含分子的子结构信息、性质信息等。因此,通过预训练,模型可以学习到某些片段的潜在表征,有助于下游任务,如结构识别任务的特征提取。这种方法在训练数据不足的情况下能增强模型的泛化性,例如在本文的模型训练中,CSR-FP用60万张化学结构图像进行训练,其在用Indigo生成的数据集的效果[SMILES序列准确率(SSA)值约为84.29%]与使用6900万张训练图像的MSE-DUDL效果(SSA值为82%)相当。并且,本文在CSRFP的解码器中引入了注意力机制,这使得模型具有更好的可解释性。最终,本文选取了OSRA、MolVec以及DECIMER作为基线方法,在两个基准数据集上进行评估,CSR-FP均取得了非常大的改进。
2 基于指纹预训练的化学结构识别
本文提出的CSR-FP的工作流示意图如图2所示。所有的数据都经过统一的预处理,处理后的数据首先进行预训练。通过一个基于指纹预训练的编码器(pre-trained encoder based on fingerprint pre-train,PE-FP)模型学习先验知识表征,然后在PE-FP模型后连接一个全连接层作为CSR-FP的编码器,学习图像的高维特征,再经过LSTM解码得到标准SMILES。
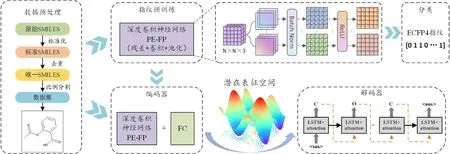
图2 CSR-FP工作流示意图Fig 2 Diagram of CSR-FP workflow
2.1 数据处理
为了获得有价值且具代表性的数据,本文选 择ZINC 20数 据 库(https://zinc20.docking.org/)作为本研究的数据源。该数据库包含超过7.5亿个可购买的化合物和超过2.3亿个可购买的可对接的化合物。如图2所示,使用RDKit化学工具包将所有原始SMILES,标准化得到标准SMILES,然后对转换后的SMILES进行去重,得到唯一SMILES,最后对数据集进行分割,按照8∶1∶1的比例获取训练数据集,并验证和测试数据集。另外,本研究中使用的化学结构图像采用Indigo化学工具包生成,并对图像进行归一化和缩放,所有的图像分辨率重置为256×256。对于PE-FP模型的标签处理,使用RDKit生成预训练化学结构图像的扩展连通性指纹(extended connectivity fingerprint,ECFP4)指纹,该指纹维度为256。而对于CSR-FP模型的标签处理,则简单地计算所有SMILES字符并添加到标签字典中。为了识别字符串的开始,结束以及填充位,在字典中添加<sos>、<eos>和<pad>标记。最终,本文选取从ZINC 20数据库中随机分层采样的600 000张化学结构图像作为CSR-FP模型训练的数据集。
为了与其他工具进行比较,本研究采用不同方法,分别为标准化学结构图像数据集(Standard-CSI)和噪声图像数据集(NI)上进行测试。两个数据集均包含10 000张化学结构图像。其中Standard-CSI数据集的图像使用Indigo工具包生成。NI数据集模拟了真实世界情况下的化学结构图像,即图像中会随机出现不同比例的图像噪声。模型的训练数据集与两个基准数据集没有重复样本,所有的基线方法在基准数据集上进行测试,该过程重复10次。
2.2 模型架构
编码-解码的思维最早在机器翻译中被广泛应用,后来由于其强大的特征空间探索能力,许多基于人工智能的方法均采用此架构。CSR-FP模型在编码器-解码器架构的基础上,融合了预训练模型PE-FP和注意力机制,增强了模型的特征提取能力和可解释性。CSR-FP和PE-RP的模型架构如图3所示,其中红色方框中的部分连接256个二分类分类器进行分子指纹学习,本研究利用长度为256的ECFP4指纹作为预训练的标签。随后,通过去除分类器并且连接一个简单的线性转换器,将得到的隐式向量转换为隐式矩阵,输入到解码器中。这里,CSR-FP模型的编码器输出的特征图大小是8×8,其通道为512。图中紫色方框中的内容为CSR-FP的解码器,解码器在每次迭代中都会生成一个SMILES字符。在训练过程中,生成字符时,模型对于前一个时间步生成的内容是可见的,而验证时则不可见。从图中还可以看出,CSR-FP的编码器中使用了多个残差连接,其是一个类ResNet的模型,通过残差连接,可以更好地解决网络退化问题。
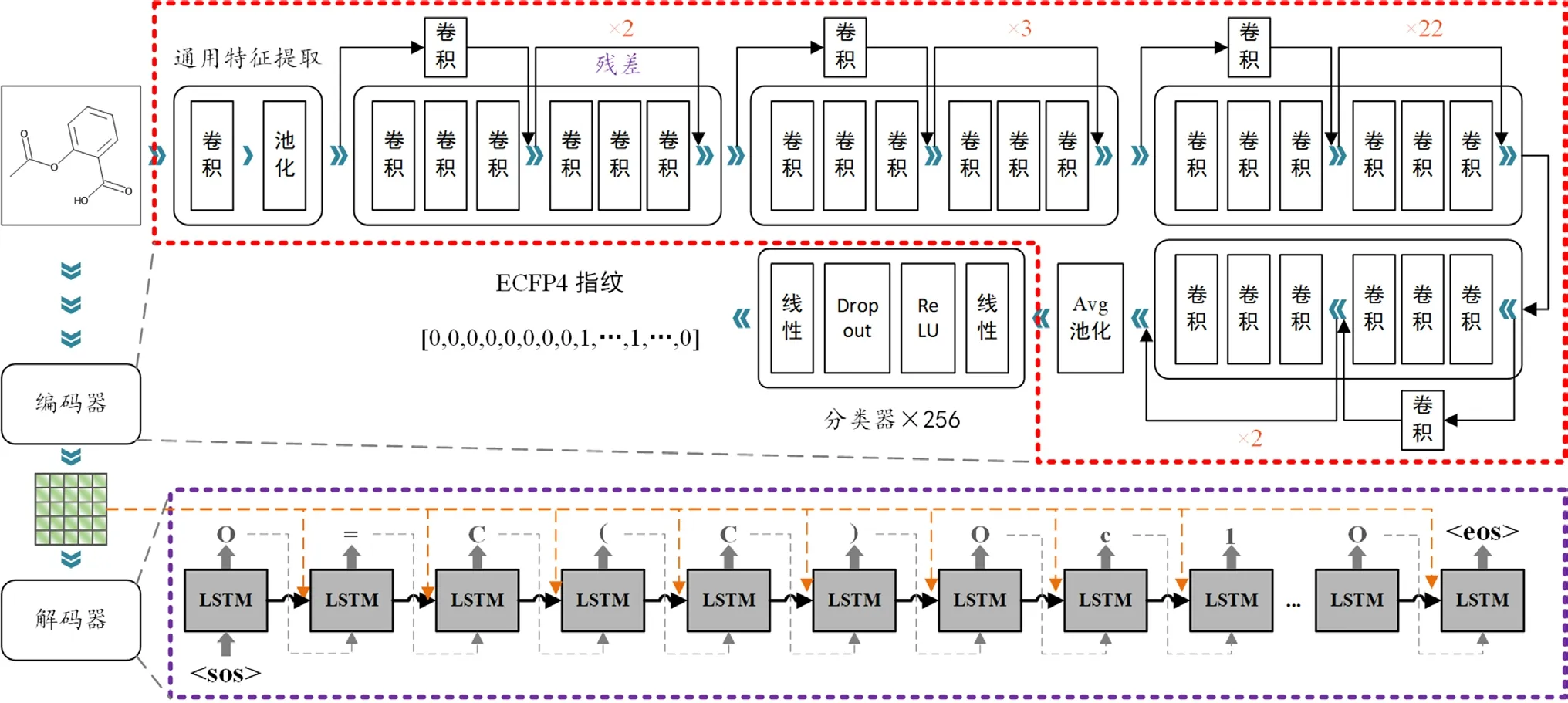
图3 模型架构示意图Fig 3 Diagram of the model architecture
假设模型预测的结果为其中N
是SMILES的长度,V
是字典的大小。模型的损失函数可以表示为:
s
个预测字符中,下标为y
的预测值。另外,CSR-FP在解码器中加入了注意力机制,在生成SMILES字符时,计算特征矩阵与前一时间步的隐含向量之间的注意力分数。这种机制可以有效克服RNN长期依赖造成的梯度消失和爆炸问题,在提升模型特征提取能力的同时,还可以增加模型的可解释性。
2.3 指纹预训练
本研究使用ECFP4指纹作为预训练的训练标签。其中,ECFP4指纹是一串由“0”和“1” 位比特组成的256长度的向量。因此,在训练过程中,设置256个分类器分别进行二分类学习。在本研究中,我们使用了100万个化合物分子进行指纹预训练。
ECFP指纹又称为Morgan指纹,是一种扩展连通性指纹。具体来说,ECFP4是以半径为2,计算每个原子的圆形区域的化学环境标识符。因此,该指纹包含了分子中多个片段的结构信息,在QSAR/QSPR任务中有着广泛的应用。
2.4 评价指标
为了更全面、更严格地评估模型的性能,本文采用了以下三个指标进行评估:
SMILES序列的准确率(SSA):在该指标下,只有当生成的SMILES序列与原始序列完全相同时才被认为是正确的结果。该指标从分子水平上宏观分析模型的识别精度。
ATS:本文使用Tanimoto系数来衡量生成的分子的Morgan指纹和原始分子的Morgan指纹之间的相似性。
Tanimoto相 似 度1.0系 数(TS@1.0):该指标是指生成的分子指纹与原始分子指纹之间Tanimoto相似度为1.0的比例。TS@1.0和ATS为从分子相似度层面上进行分析,这些指标可以为下游任务,例如分子性质的预测等奠定基础。
2.5 注意力机制
一般来说,图像中包含了多个不同的内容,化学结构图像也不例外,其中不同的原子或化学键对应的图像块可以看成是不同的区域。因此,如果在生成SMILES字符时能准确估计图像中某一区域的重要性,那么模型在性能上将会得到质的提升。并且,在生成文本内容时,每个字符之间都有一定的联系,因此,在生成当前字符时,应该整合已经生成的文本信息,即当前时间步S
之前的迭代(S
,S
,…,S
)生成的SMILES字符信息。在解码器的每个时间步中,模型输出一个隐藏向量h
和一个单元向量c
,其中初始向量h
和c
是将编码器的特征向量输入全连接层得到的。每个时间步中注意力分数的计算方式可以简单地形式化为:
L
是一个线性转换,F
是特征矩阵,h
是解码器第t
个时间步的隐式向量。3 结果
3.1 数据集分析
为了提升模型的泛化能力,在进行指纹预训练时,多样性更高、应用域更广的数据对于提升预训练模型的学习能力是非常重要的。针对该问题,我们在采集预训练样本时,利用分子骨架多样性对分子多样性进行度量,采集骨架多样性高的样本进行训练。预训练数据集的分子骨架分析以及原子数目分布结果,见图4。
由图4A可知,分子骨架数量平均值为1.154,且数据主要分布在最小值1处。小部分样本分布在2和3中。从图4C中可以看出,小于3的分子骨架数量占比为97.76%。这些数据说明了该预训练数据集的分子结构多样性非常丰富。从图4B可以看出,原子数目大部分集中在20~40,比较符合高成药性化合物的特点。总而言之,通过该数据集学习到的化学空间表征是更有效的。

图4 预训练数据集统计图Fig 4 Statistical diagram of pre-trained dataset
3.2 基线对比实验
为了验证本文提出的模型的有效性,本文将CSR-FP模型与一些现有的模型和工具进行了比较。其中,基于规则的工具有OSRA和MolVec,基于深度学习的工具有DECIMER和CSR-FP。评估指标的统计数据见表1。
表1 模型性能对比结果
Tab 1 Model performance
数据集 方法 SSA/% ATS/% TS@1.0/%Standard-CSIOSRA 22.05±0.22 54.15±0.36 36.92±0.62 Standard-CSI MolVec 17.18±0.41 50.59±0.24 31.18±0.23 Standard-CSIDECIMER 38.19±0.34 87.52±0.21 47.54±0.22 Standard-CSICSR-FP 84.29±0.12 92.14±0.13 87.49±0.16 NI OSRA 19.52±0.46 51.41±0.37 32.02±0.19 NI MolVec 8.714±0.37 34.79±0.19 18.01±0.35 NI DECIMER 17.42±0.27 68.46±0.29 22.16±0.38 NI CSR-FP 82.19±0.17 88.19±0.18 84.86±0.21
对于以上三个评估数据,我们主要关注SSA值。从表1中可以看出,CSR-FP在两个数据集上均取得了具有竞争力的结果。具体来说,两个基于规则的工具在两个数据集上的效果均不理想,例如OSRA和MolVec在Standard-CSI数据集上的SSA值只有22.05%和17.18%,而DECIMER和CSR-FP则为38.19%和84.29%。这说明相对于基于深度学习的方法,基于规则的方法泛化能力较差。而对于带有噪声的图像,前三者的识别精度均低于20%,可能是因为基于规则的方法没有加入降噪的规则。DECIMER则是直接使用一个特征提取器而没有和解码器进行联动学习,这会大大降低模型的特征学习能力。总体来说,CSR-FP通过融合分子指纹预训练以及注意力机制,大大提升了模型的特征提取能力。
3.3 可视化结果
在CSR-FP模型中,编码器的输出为一个512通道的8×8特征图,在解码器生成字符时,模型计算特征图与隐式向量的注意力分数。因此,本文随机选取了一个正确识别的分子并将其每个字符生成时模型对64个图像块的注意力分数进行了可视化。如图5所示,第一个和最后一个子图分别表示原始的分子图片与所有字符叠加的注意力权重图。从最后一个子图可以看出,模型能够大致识别整个图片中的分子区域。从中间的字符权重子图也可以看出,模型关注的区域与相关原子的位置有所重合。虽然某些字符的关注权重可能有所偏差,但是模型可以利用注意力信息,整合上下文信息以提升解码能力。未来,我们将通过扩大特征图以细化模型对分子图像块的信息抽取能力。

图5 注意力机制示例图Fig 5 Example diagram of attentional mechanisms
4 结论
针对CSR,本文构建了一个基于编码器-解码器架构的模型CSR-FP。该模型能较好地学习分子图像的表征,并且具备良好的可解释性。两个基准数据集测试结果表明该模型在目前已有的工具和方法中取得了具有竞争力的效果。具体来说,本文的贡献主要包括三个方面:
第一,构建了一个基于编码器-解码器架构的模型CSR-FP,该模型能较好地学习分子图像空间表征,与其他工具及方法相比取得了具有竞争力的效果。该工作可在如下链接获取:https://github.com/Jiacai-Yi/CSR-FP。
第二,采用了深度学习的方法,融合了分子指纹知识,即模型在学习分子图像空间时能结合
预训练学习到的分子结构信息和性质信息,对于生成更有效的空间表征具有指导意义。
第三,解码器融合了注意力机制,使模型在解码每个SMILES字符时都与编码形成的特征图做注意力计算,不仅提升了模型的特征提取能力,同时让模型具有更好的可解释性。
后续工作中,我们将深入探讨化学结构图像内在特征对模型性能的影响,并且构建与该任务更匹配的模型。并且,由于我们最终的目标是在大量文献中分割并识别准确的分子结构,因此,未来还需要在排除文本干扰、准确识别R-基团以及识别复杂的立体异构分子等问题上进行深入研究。
