新型2-三氟甲基-4-氨基喹啉衍生物的合成及抗肿瘤活性研究
2022-03-25吕梦凡曾晓萍孟雪玲徐广灿徐必学
吕梦凡, 余 佳, 曾晓萍, 孟雪玲, 徐广灿*, 徐必学*
(1. 贵州中医药大学,药学院,贵州 贵阳 550025; 2. 贵州医科大学 省部共建药用植物功效与利用国家重点实验室,贵州 贵阳 550014; 3. 贵州省中国科学院天然产物化学重点实验室,贵州 贵阳 550014)
Chart 1
据国际癌症研究中心(International Agency for Research on Cancer, IARC)发布的最新一期《全球癌症统计2020》显示,2020年全球新诊断癌症为1929万例,癌症死亡人数超过2018年的960万人,预计将达1000万人,其中肺癌、结直肠癌、肝癌及胃癌是全球死亡率最高的癌症[1-2]。目前,治疗癌症的方法主要包括手术、放疗、化疗、免疫治疗等,其中化疗始终是最主要的干预措施[3]。但现有化疗药物往往存在毒副作用大、耐受性差等缺点,因此寻找高疗效、低毒副作用的抗肿瘤药物显得尤为迫切[4]。
含氮杂环化合物一直是药物设计和开发的热点,喹啉作为一类重要的氮杂环化合物,也是许多天然产物或合成的生物活性化合物的母核,因具有高效且多样的生物活性,在医药领域得到广泛的应用,例如:抗肿瘤[5-6]、抗疟疾[7]、调节免疫[8]、抗菌[9-10]、抗炎症[11]、抗糖尿病等[12]。近年来,多种以喹啉为母体的抗肿瘤药物正处于临床研究阶段或已上市,例如安罗替尼(Anlotinib)[13]、乐伐替尼(Lenvatinib)[14]及卡博替尼(Cabozantinib)[15]等已经上市的药物,德立替尼(Lucitanib)[16]等处于二期临床的喹啉化合物和宁格替尼(Ningetinib)[17]等处于一期临床的喹啉化合物,均为多靶点络氨酸酶抑制剂(Chart 1)。
喹啉母核结构上取代基的变化往往对其药效及生物活性具有一定影响,其中三氟甲基是一类十分重要的化学基团,由于其具有强吸电子性、化学稳定性、物理亲脂性等性质,使得含有三氟甲基的化合物已经在医药方面得到了广泛的应用,将三氟甲基引入到喹啉单元中,可以显著提高其代谢稳定性、亲脂性、生物利用度以及结合亲和力[18-20]。本课题组前期开展了2-三氟甲基-3-苯甲酰胺基喹啉衍生物的合成及抗肿瘤活性研究[21],为进一步探索该类化合物在抗肿瘤方面的应用,本文设计并合成了一系列2-三氟甲基-4-氨基喹啉衍生物(5a~5e、6、7a~7d),合成路线见Scheme 1,并对合成所得目标化合物均进行了体外抗肿瘤活性研究,以期寻到具有良好抗肿瘤活性的目标化合物,为进一步研发新型抗肿瘤药物奠定基础。
1 实验部分
1.1 仪器与试剂
XT-4型熔点仪;Bruker Avance NEO 600 MHz型核磁共振仪(DMSO-d6或CDCl3为溶剂,TMS为内标);Hewlett-Packard HP-5793型质谱仪。
所用试剂均为分析纯或化学纯。
1.2 合成
(1) 化合物2的合成
称取多聚磷酸38.01 g(112.5 mmol)于100 mL反应瓶中,用氩气置换3次,于100 ℃搅拌0.5 h后,依次缓慢注入对氟苯胺4.27 mL(45.0 mmol)、三氟乙酰乙酸乙酯6.58 mL(45.0 mmol),并继续于100 ℃搅拌3.5 h至反应完全。将反应液分散至300 mL水中,析出大量固体,过滤,滤饼用水洗涤至中性,于80 ℃减压烘干得化合物2粗品7.86 g,未经进一步纯化直接用于下一步反应。

Scheme 1
(2) 化合物3的合成
称取化合物21.30 g(5.62 mmol)于25 mL反应瓶中,加入10 mL DMF使其完全溶解;用氩气置换3次,搅拌下于100 ℃缓慢注入三氯氧磷1.54 mL(16.9 mmol),并继续于100 ℃搅拌4.5 h至反应完全。待反应液冷却至室温后,将其分散于乙酸乙酯与水中,萃取,有机层依次用饱和食盐水洗涤,无水MgSO4干燥,过滤,滤液减压浓缩,残余物经硅胶柱层析(洗脱剂:石油醚)纯化得白色固体状化合物31.24 g,收率88.5%;1H NMR(600 MHz, CDCl3)δ: 8.26(dd,J=9.2 Hz, 5.2 Hz, 1H), 7.90(dd,J=9.1 Hz, 2.7 Hz, 1H), 7.85(s, 1H), 7.65(ddd,J=9.4 Hz, 7.9 Hz, 2.8 Hz, 1H);13C NMR(151 MHz, CDCl3)δ: 162.43(d,J=254.2 Hz), 147.17(q,J=35.7 Hz), 145.02, 143.80(d,J=6.2 Hz), 133.48(d,J=9.8 Hz), 128.35(d,J=10.7 Hz), 122.27(d,J=26.0 Hz), 120.92(q,J=275.3 Hz), 117.88, 108.07(d,J=24.8 Hz)。
(3) 化合物4a~4c的合成(以4a为例)
称取化合物3500 mg(2.00 mmol)、 3,4,5-三甲氧基苯胺440 mg(2.00 mmol)于25 mL反应瓶中,加入8 mL异丙醇将其完全溶解,用氩气置换3次,搅拌下于80 ℃注入浓盐酸0.17 mL(2.0 mmol),并继续于80 ℃搅拌2.5 h至反应完全。待反应液冷却至室温后,抽滤反应体系中析出的固体,滤饼于60 ℃减压烘干得黄色固体状化合物4a纯品635.9 mg,收率80.1%;1H NMR(600 MHz, DMSO-d6)δ: 9.34(s, 1H), 8.31(dd,J=10.7 Hz, 2.8 Hz, 1H), 8.07(dd,J=9.3 Hz, 5.6 Hz, 1H), 7.75(ddd,J=9.3 Hz, 8.0 Hz, 2.8 Hz, 1H), 7.11(s, 1H), 6.74(s, 2H), 3.78(s, 6H), 3.71(s, 3H);13C NMR(151 MHz, DMSO-d6)δ: 160.54(d,J=245.5 Hz), 154.02, 150.84(d,J=4.5 Hz), 147.30(q,J=32.3 Hz), 145.39, 135.57, 135.29, 133.04(d,J=9.0 Hz), 122.22(q,J=275.4 Hz), 121.15(d,J=25.0 Hz), 120.58(d,J=9.7 Hz), 106.75(d,J=23.8 Hz), 102.08, 96.62, 60.65, 56.47。
用类似的方法合成化合物4b~4c。
4b: 白色固体,收率91.5%;1H NMR(600 MHz, CDCl3)δ: 8.15(dd,J=9.3 Hz, 5.5 Hz, 1H), 7.57(dd,J=9.6 Hz, 2.7 Hz, 1H), 7.53(ddd,J=9.3 Hz, 7.8 Hz, 2.6 Hz, 1H), 7.37(t,J=8.1 Hz, 1H), 7.31(s, 1H), 6.90(dd,J=8.0 Hz, 2.0 Hz, 1H), 6.84(t,J=2.3 Hz, 1H), 6.82(dd,J=8.3 Hz, 2.4 Hz, 1H), 6.66(s, 1H), 3.84(s, 3H);13C NMR(151 MHz, CDCl3)δ: 161.07(d,J=250.3 Hz), 160.98, 148.90(d,J=5.1 Hz), 148.27(q,J=33.6, Hz), 145.24, 139.81, 133.51(d,J=8.9 Hz), 130.84, 121.64(q,J=275.3 Hz), 120.63(d,J=25.5 Hz), 120.41(d,J=8.7 Hz), 115.23, 111.22, 108.93, 103.89(d,J=23.5 Hz), 98.33, 55.44。
4c: 黄色固体,收率82.4%;1H NMR(600 MHz, DMSO-d6)δ: 9.57(s, 1H), 8.42(dd,J=10.7 Hz, 2.8 Hz, 1H), 8.07(dd,J=9.3 Hz, 5.6 Hz, 1H), 7.75(ddd,J=9.2 Hz, 8.0 Hz, 2.8 Hz, 1H), 7.48(d,J=7.1 Hz, 2H), 7.41(t,J=7.5 Hz, 2H), 7.37~7.31(m, 3H), 7.15(d,J=8.8 Hz, 2H), 6.80(s, 1H), 5.14(s, 2H);13C NMR(151 MHz, DMSO-d6)δ: 160.48(d,J=244.6 Hz), 156.88, 151.73(d,J=4.8 Hz), 147.23(d,J=33.0 Hz), 145.29, 137.42, 132.93(d,J=9.7 Hz), 132.02, 128.92, 128.38, 128.30, 126.86, 122.20(q,J=275.3 Hz), 121.13(d,J=25.1 Hz), 120.27(d,J=9.6 Hz), 116.40, 106.83(d,J=23.7 Hz), 95.39, 69.99。
(4) 化合物4d的合成
称取化合物3150 mg(0.60 mmol)、 2-氨基嘧啶69 mg(0.72 mmol)于10 mL反应管中,加入3 mL DMF使其完全溶解,加NaH 72 mg(1.8 mmol),用氩气置换3次,于80 ℃搅拌18 h至反应完全。待反应液冷却至室温后,将其分散于乙酸乙酯与水中,萃取,有机层依次用饱和食盐水洗涤,无水MgSO4干燥,过滤,滤液减压浓缩,残余物经硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=1/20,V/V)纯化得白色固体状化合物4d139.9 mg,收率75.5%;1H NMR(600 MHz, CDCl3)δ: 9.02(s, 1H), 8.61(d,J=4.7 Hz, 2H), 8.22(dd,J=9.3 Hz, 5.5 Hz, 1H), 8.00(s, 1H), 7.65(dd,J=9.7 Hz, 2.6 Hz, 1H), 7.57(ddd,J=9.8 Hz, 7.8 Hz, 2.6 Hz, 1H), 6.99(t,J=4.8 Hz, 1H);13C NMR(151 MHz, CDCl3)δ: 161.44(d,J=251.6 Hz), 159.20, 158.26, 148.36(q,J=33.7 Hz), 144.92, 143.60(d,J=5.0 Hz), 133.90(d,J=9.0 Hz), 121.73(q,J=275.3 Hz), 120.77(d,J=8.9 Hz), 120.55(d,J=25.9 Hz), 115.14, 104.25, 103.75(d,J=23.5 Hz)。
(5) 化合物5a~5d的合成(以5a为例)
称取化合物4a80 mg(0.20 mmol)于10 mL反应管中,加入2 mL DMF使其完全溶解,加入NaH 20 mg(0.50 mmol),用氩气置换3次,搅拌下缓慢注入碘甲烷31 μL(0.50 mmol),反应3 h至终点。将反应液分散于乙酸乙酯与水中,萃取,有机层依次用饱和食盐水洗涤,无水MgSO4干燥,过滤,滤液减压浓缩,残余物经硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=1/20,V/V)纯化得白色固体状化合物5a78.9 mg,收率95.4%, m.p.129~132 ℃;1H NMR(600 MHz, CDCl3)δ: 8.13(dd,J=9.2 Hz, 5.6 Hz, 1H), 7.42(ddd,J=9.2 Hz, 7.6 Hz, 2.8 Hz, 1H), 7.32(s, 1H), 7.23(dd,J=10.4 Hz, 2.8 Hz, 1H), 6.20(s, 2H), 3.84(s, 3H), 3.71(s, 6H), 3.50(s, 3H);13C NMR(151 MHz, CDCl3)δ: 160.28(d,J=249.4 Hz), 154.55(d,J=5.0 Hz), 154.08, 148.08(q,J=33.6 Hz), 145.97, 145.23, 135.32, 133.16(d,J=9.0 Hz), 124.65(d,J=9.9 Hz), 121.67(q,J=275.5 Hz), 120.38(d,J=26.1 Hz), 108.91(d,J=24.7 Hz), 106.82, 100.41, 61.08, 56.26, 43.32;19F NMR(565 MHz, CDCl3)δ: -67.71, -110.03; MS(ESI)m/z: 433.3{[M+Na]+}(Calcd for C20H18F4N2NaO3, 433.12)。
用类似的方法合成化合物5b~5d。
5b: 黄色固体,收率89.1%, m.p.90~92 ℃;1H NMR(600 MHz, CDCl3)δ: 8.16(dd,J=9.3 Hz, 5.5 Hz, 1H), 7.44(ddd,J=9.3 Hz, 7.8 Hz, 2.9 Hz, 1H), 7.39(s, 1H), 7.30~7.24(m, 1H), 7.19(t,J=8.1 Hz, 1H), 6.63(dd,J=8.4 Hz, 2.4 Hz, 1H), 6.51(dd,J=8.0 Hz, 2.2 Hz, 1H), 6.49(t,J=2.4 Hz, 1H), 3.74(s, 3H), 3.51(s, 3H);13C NMR(151 MHz, CDCl3)δ: 160.79, 160.58(d,J=249.6 Hz), 154.83(d,J=6.0 Hz), 150.20, 148.18(q,J=33.7 Hz), 146.09, 133.23(d,J=8.9 Hz), 130.46, 125.42(d,J=9.3 Hz), 121.62(q,J=275.5 Hz), 120.56(d,J=26.0 Hz), 113.75, 108.89, 108.70(d,J=8.6 Hz), 108.50, 107.60, 55.34, 42.57;19F NMR(565 MHz, CDCl3)δ: -67.71, -109.99; MS(ESI)m/z: 351.2{[M+H]+}(Calcd for C18H15F4N2O, 351.11)。
5c: 黄色固体,收率91.3%, m.p.80~85 ℃;1H NMR(600 MHz, CDCl3)δ: 8.11(dd,J=9.3 Hz, 5.6 Hz, 1H), 7.46~7.30(m, 6H), 7.26(s, 1H), 7.15(dd,J=10.7 Hz, 2.8 Hz, 1H), 6.96(d,J=9.1 Hz, 2H), 6.93(d,J=9.0 Hz, 2H), 5.05(s, 2H), 3.47(s, 3H);13C NMR(151 MHz, CDCl3)δ: 159.96(d,J=248.1 Hz), 156.05, 154.81(d,J=5.0 Hz), 148.05(q,J=33.9 Hz), 146.03, 142.97, 136.69, 133.07(d,J=9.5 Hz), 128.64, 128.11, 127.55, 124.74, 124.08(d,J=9.4 Hz), 121.75(q,J=275.3 Hz), 120.02(d,J=26.0 Hz), 116.20, 109.34(d,J=24.5 Hz), 105.34, 70.39, 43.70;19F NMR(565 MHz, CDCl3)δ: -66.74, -112.49; MS(ESI)m/z: 449.5{[M+Na]+}(Calcd for C24H18F4N2NaO, 449.13)。
5d: 黄色固体,收率44.8%, m.p.77~80 ℃;1H NMR(600 MHz, CDCl3)δ: 8.35(d,J=4.8 Hz, 2H), 8.29(dd,J=9.3 Hz, 5.3 Hz, 1H), 7.68(s, 1H), 7.57(ddd,J=9.2 Hz, 7.9 Hz, 2.8 Hz, 1H), 7.36(dd,J=9.3 Hz, 2.8 Hz, 1H), 6.73(t,J=4.8 Hz, 1H), 3.65(s, 3H);13C NMR(151 MHz, CDCl3)δ: 161.84(d,J=252.0 Hz), 161.71, 157.99, 152.68(d,J=5.8 Hz), 148.30(q,J=34.8 Hz), 146.30, 133.63(d,J=8.9 Hz), 128.12(d,J=9.6 Hz), 121.42(d,J=27.0 Hz), 121.36(q,J=275.3 Hz), 116.04, 112.49, 107.36(d,J=23.6 Hz), 38.75;19F NMR(565 MHz, CDCl3)δ: -67.42, -108.86; MS(ESI)m/z: 323.1{[M+H]+}(Calcd for C15H11F4N4, 323.09)。
(6) 化合物5e的合成
称取化合物3100 mg(0.40 mmol)、N-甲基对硝基苯胺67 mg(0.44 mmol)于10 mL反应管中,加入10 mL DMF使其溶解,加NaH 48 mg(1.2 mmol),用氩气置换3次,于80 ℃搅拌反应12 h至反应完全。待反应液冷却至室温后,将其分散于乙酸乙酯与水中,萃取,有机层依次用饱和食盐水洗涤,无水MgSO4干燥,过滤,滤液减压浓缩,残余物经硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=1/10,V/V)纯化得黄色固体状化合物5e70.0 mg,收率47.8%, m.p.116~123 ℃;1H NMR(600 MHz, CDCl3)δ: 8.33(dd,J=9.3 Hz, 5.3 Hz, 1H), 8.12(d,J=9.3 Hz, 2H), 7.66(s, 1H), 7.63(ddd,J=9.3 Hz, 7.8 Hz, 2.8 Hz, 1H), 7.35(dd,J=9.1 Hz, 2.8 Hz, 1H), 6.70(d,J=9.3 Hz, 2H), 3.59(s, 3H);13C NMR(151 MHz, CDCl3)δ: 162.11(d,J=254.2 Hz), 153.05(d,J=6.2 Hz), 152.65, 148.69(q,J=35.9 Hz), 146.61, 140.41, 134.11(d,J=9.8 Hz), 127.10(d,J=9.8 Hz), 125.90, 121.16(q,J=275.3 Hz), 122.09(d,J=26.2 Hz), 115.40, 114.26, 107.09(d,J=23.6 Hz), 40.88;19F NMR(565 MHz, CDCl3)δ: -67.47, -106.86; MS(ESI)m/z: 364.0{[M-H]-}(Calcd for C17H10F4N3O2, 364.07)。
(7) 化合物6的合成
称取化合物5c51 mg(0.12 mmol)于25 mL反应瓶中,加入7 mL甲醇使其完全溶解,再加Pd/C(10%)3.8 mg(0.004 mmol),浓盐酸0.04 mL(0.48 mmol),用氢气置换3次,室温反应24 h至反应完全。用300~400目硅胶吸附抽滤,滤液减压浓缩,残余物经硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=1/5,V/V)纯化得黄色固体状化合物638.0 mg,收率94.5%, m.p.172~174 ℃;1H NMR(600 MHz, CDCl3)δ: 8.69(s, 1H), 8.03(dd,J=9.3 Hz, 5.6 Hz, 1H), 7.31(ddd,J=9.3 Hz, 7.6 Hz, 2.9 Hz, 1H), 7.16(s, 1H), 7.10(dd,J=11.0 Hz, 2.8 Hz, 1H), 6.86(d,J=8.8 Hz, 2H), 6.78(d,J=8.8 Hz, 2H), 3.42(s, 3H);13C NMR(151 MHz, CDCl3)δ: 159.59(d,J=248.2 Hz), 154.95, 154.80(d,J=5.0 Hz), 147.84(q,J=32.4 Hz), 145.91, 141.58, 132.78(d,J=9.7 Hz), 125.35, 123.69(d,J=9.6 Hz), 121.78(q,J=275.3 Hz), 119.76(d,J=26.0 Hz), 116.86, 109.59(d,J=24.9 Hz), 104.15, 43.95;19F NMR(565 MHz, CDCl3)δ: -67.71, -111.14; MS(ESI)m/z: 359.0{[M+Na]+}(Calcd for C17H12F4N2NaO, 359.08)。
(8) 化合物7a~7d的合成(以7a为例)
称取化合物6100 mg(0.297 mmol)、 K2CO3164 mg(1.19 mmol)于10 mL反应管中,加入3 mL DMF使其完全溶解,用氩气置换3次,搅拌下于80 ℃缓慢注入1-溴-3-氯丙烷38 μL(0.39 mmol),并继续于80 ℃搅拌20 h至反应完全。待反应液冷却至室温后,将其分散于乙酸乙酯与水中,萃取,有机层依次用饱和食盐水洗涤,无水MgSO4干燥,过滤,滤液减压浓缩,残余物经硅胶柱层析(洗脱剂:乙酸乙酯/石油醚=1/15,V/V)纯化得白色固体状化合物7a122.6 mg,收率99.8%, m.p.87~90 ℃;1H NMR(600 MHz, CDCl3)δ: 8.10(dd,J=9.2 Hz, 5.6 Hz, 1H), 7.37(ddd,J=9.3 Hz, 7.7 Hz, 2.9 Hz, 1H), 7.26(s, 1H), 7.15(dd,J=10.7 Hz, 2.8 Hz, 1H), 6.96(d,J=8.9 Hz, 2H), 6.85(d,J=9.0 Hz, 2H), 4.10(t,J=5.9 Hz, 2H), 3.75(t,J=6.3 Hz, 2H), 3.47(s, 3H), 2.27~2.20(m, 2H);13C NMR(151 MHz, CDCl3)δ: 159.96(d,J=248.2 Hz), 155.99, 154.81(d,J=6.0 Hz), 148.04(q,J=33.8 Hz), 146.02, 142.96, 133.07(d,J=9.8 Hz), 124.76, 124.08(d,J=9.5 Hz), 121.75(q,J=275.4 Hz), 120.02(d,J=26.0 Hz), 115.79, 109.32(d,J=24.1 Hz), 105.35, 64.61, 43.72, 41.46, 32.25;19F NMR(565 MHz, CDCl3)δ: -67.74, -110.63; MS(ESI)m/z: 435.0{[M+Na]+}(Calcd for C20H17ClF4N2NaO, 435.09)。
用类似的方法合成化合物7b~7d。
7b: 黄色固体,收率87.4%, m.p.59~60 ℃;1H NMR(600 MHz, CDCl3)δ: 8.08(dd,J=9.2 Hz, 5.6 Hz, 1H), 7.36(ddd,J=9.3 Hz, 7.8 Hz, 2.8 Hz, 1H), 7.23(s, 1H), 7.13(dd,J=10.7 Hz, 2.8 Hz, 1H), 6.94(d,J=8.9 Hz, 2H), 6.84(d,J=8.9 Hz, 2H), 3.99(t,J=6.3 Hz, 2H), 3.45(s, 3H), 2.52(t,J=7.4 Hz, 2H), 2.30(s, 6H), 2.01~1.95(m, 2H);13C NMR(151 MHz, CDCl3)δ: 159.89(d,J=248.2 Hz), 156.30, 154.81(d,J=5.0 Hz), 148.01(q,J=33.0 Hz), 146.00, 142.72, 133.03(d,J=8.8 Hz), 124.81, 124.01(d,J=9.9 Hz), 121.75(q,J=275.4 Hz), 119.96(d,J=26.0 Hz), 115.77, 109.36(d,J=24.7 Hz), 105.14, 66.39, 56.24, 45.22, 43.75, 27.22;19F NMR(565 MHz, CDCl3)δ: -67.74, -110.74; MS(ESI)m/z: 422.1{[M+H]+}(Calcd for C22H24F4N3O, 422.19)。
7c: 黄色固体,收率89.2%, m.p.34~35 ℃;1H NMR(600 MHz, CDCl3)δ: 8.09(dd,J=9.2 Hz, 5.7 Hz, 1H), 7.36(ddd,J=9.2 Hz, 7.6 Hz, 2.8 Hz, 1H), 7.24(s, 1H), 7.13(dd,J=10.8 Hz, 2.8 Hz, 1H), 6.95(d,J=8.9 Hz, 2H), 6.87(d,J=8.9 Hz, 2H), 4.04(t,J=5.7 Hz, 2H), 3.46(s, 3H), 2.73(t,J=5.7 Hz, 2H), 2.34(s, 6H);13C NMR(151 MHz, CDCl3)δ: 159.90(d,J=249.2 Hz), 156.18, 154.80(d,J=5.0 Hz), 148.01(q,J=35.2, Hz), 146.01, 142.79, 133.03(d,J=9.7 Hz), 124.76, 124.02(d,J=9.8 Hz), 121.75(q,J=275.2 Hz), 119.98(d,J=26.0 Hz), 115.85, 109.36(d,J=24.1 Hz), 105.17, 66.27, 58.30, 45.93, 43.72;19F NMR(565 MHz, CDCl3)δ: -67.75, -110.72; MS(ESI)m/z: 408.4{[M+H]+}(Calcd for C21H22F4N3O, 408.17)。
7d: 黄色油状液体,收率86.9%;1H NMR(600 MHz, CDCl3)δ: 8.09(dd,J=9.3 Hz, 5.6 Hz, 1H), 7.36(ddd,J=9.3 Hz, 7.7 Hz, 2.8 Hz, 1H), 7.24(s, 1H), 7.13(dd,J=10.8 Hz, 2.8 Hz, 1H), 6.95(d,J=8.9 Hz, 2H), 6.85(d,J=9.0 Hz, 2H), 4.03(t,J=6.2 Hz, 2H), 3.46(s, 3H), 2.87(t,J=6.3 Hz, 2H), 2.64(q,J=7.1 Hz, 4H), 1.07(t,J=7.1 Hz, 6H);13C NMR(151 MHz, CDCl3)δ: 159.89(d,J=248.4 Hz), 156.21, 154.80(d,J=5.1 Hz), 148.02(q,J=33.9 Hz), 146.01, 142.72, 133.02(d,J=9.6 Hz), 124.80, 124.01(d,J=9.9 Hz), 121.75(q,J=275.4 Hz), 119.97(d,J=25.1 Hz), 115.81, 109.37(d,J=24.8 Hz), 105.12, 66.85, 51.69, 47.87, 43.74, 11.79;19F NMR(565 MHz, CDCl3)δ: -67.75, -110.72; MS(ESI)m/z: 436.4{[M+H]+}(Calcd for C23H26F4N3O, 436.20)。
1.3 抗肿瘤活性测试
以阿霉素和紫杉醇作为阳性对照,采用MTT法[22]测试目标化合物5a~5e、6和7a~7d对前列腺癌细胞(PC3、 LNCaP)以及慢性髓系白血病细胞(K562)的体外抑制活性。分别取上述3种处在对数期的癌细胞,接种在96孔板中,培养24 h,用DMSO溶解目标化合物、阿霉素和紫杉醇,使用无血清的培养基来稀释给药,保证每孔板的化合物终浓度为5 μmol·L-1,继续培养48 h,再加入20 μL MTT溶液(5 mg·mL-1)继续培养4 h,弃去上清液,加入150 μL DMSO,避光、低速震荡至完全溶解。在490 nm波长下使用酶联免疫检测仪测量吸光度(OD)值,每组重复3次,记录结果并计算抑制率。
2 结果与讨论
2.1 合成
以化合物1为起始原料,通过与三氟乙酰乙酸乙酯发生环合反应生成化合物2;2经氯代反应得到化合物3;3和苯胺类衍生物经偶联反应得到化合物4a~4d与5e;4a~4d进行N-甲基化反应得到化合物5a~5d;5c经Pd/C还原脱苄基得到化合物6;6经亲核取代反应得到7a~7d。其中在化合物4a的合成过程中,无需柱层析纯化,反应液直接抽滤即可获得纯品,操作简单且收率高。
2.2 抗肿瘤活性测试
由表1可知,(1)合成所得所有目标化合物对PC3、 LNCaP以及K562细胞均具有一定的抑制作用,在5 μmol·L-1浓度下,化合物5b、5c及6对PC3和K562细胞、化合物7a对K562细胞的抑制率均超过50%。(2)通过化合物5b、5c、6与5e结构比较可知,当苯环上为单取代时,供电子基的引入对目标化合物体外抗肿瘤活性有利。(3)通过化合物5a与5b结构比较可知,当苯环上均为供电子基取代时,单取代优于多取代。(4)通过化合物7b~7d与6结构比较可知,当苯环上的羟基被N,N-二甲氨基氯丙烷、二甲氨基氯乙烷及2-二乙氨基氯乙烷取代时,不利于目标化合物的体外抗肿瘤活性。
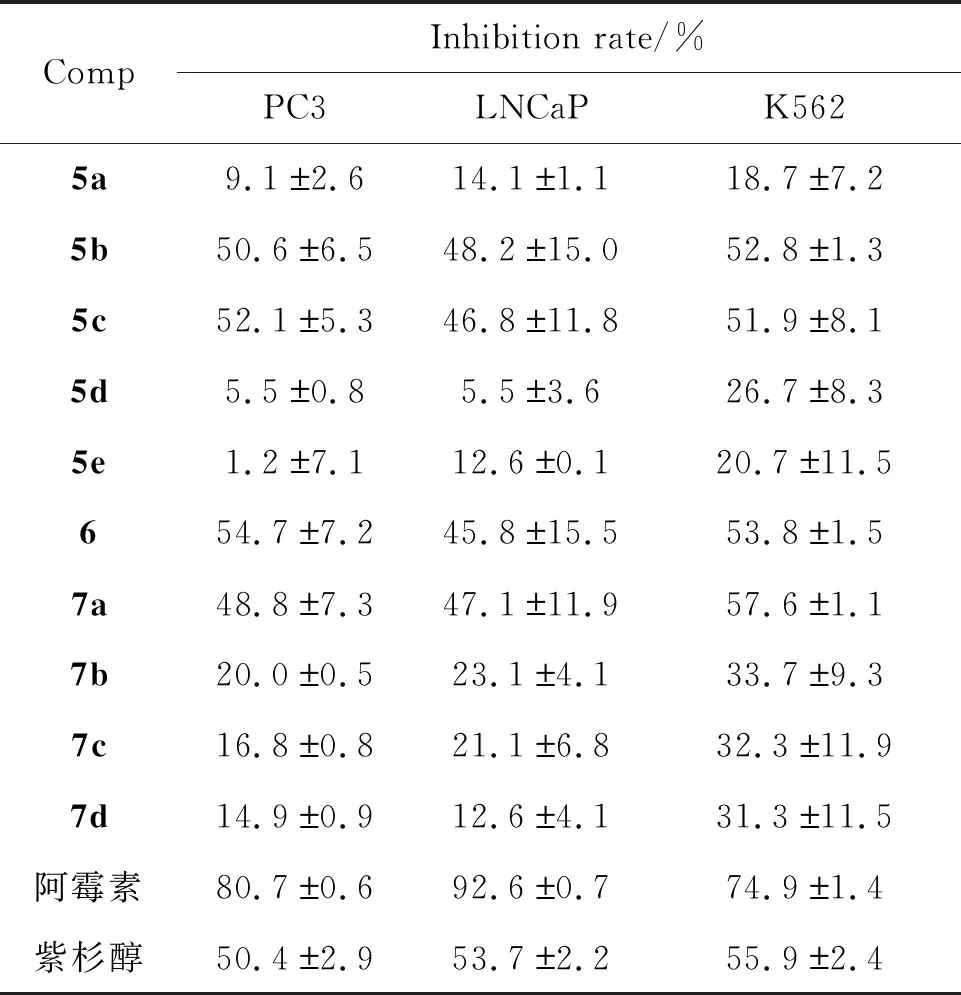
表1 化合物的体外抗肿瘤活性
设计并合成了一系列2-三氟甲基-4-氨基喹啉衍生物。采用MTT法对所合成的10个目标化合物进行了体外抗肿瘤活性测试。结果显示,大部分衍生物对PC3、 LNCaP以及K562细胞具有一定的体外抑制作用,其中化合物5b、5c、6和7a对3种细胞株均有良好的抑制作用,在5 μmol·L-1浓度下,化合物5b、5c及6对PC3细胞的抑制率(50.6%、 52.1%、 54.7%)优于阳性对照药紫杉醇(50.4%)。在5 μmol·L-1浓度下,化合物7a对K562细胞的抑制率(57.6%)强于紫杉醇(55.9%)。在此研究基础上,后续可在喹啉环5、 7及8-位进行多样化修饰,从而进一步探讨取代基的变化对喹啉衍生物抗肿瘤活性的影响。
