新型含吡唑香豆素结构的羟胺类荧光探针的合成及其在醛类化合物检测中的应用
2022-03-25张文清杜传元朱芳芳胡晓松刘亚群
朱 灏, 张文清, 杜传元, 朱芳芳, 胡晓松, 刘亚群
(武汉理工大学 化学化工与生命科学学院,湖北 武汉 430070)
醛类化合物广泛存在于食物[1-6]、饮料[7-12]、生态环境[13-16]和生物系统中[17-22],它们的含量对于人体健康和人类的生活质量有深远影响。以食品工业为例,醛类化合物的含量是食品质量评价体系的重要指标之一。低含量的醛类物质的存在能提供食品令人愉悦的风味,而当醛类物质的含量过高时会加速食品的变质。糠醛(F)、 5-甲基糠醛(5-MF)、 5-羟甲基糠醛(5-HMF)广泛存在于各种食品中,其含量是其新鲜度和质量的重要指标[2,5,10,12],因此醛类物质的定量分析和检测具有重要的研究意义。目前已经有基于紫外-可见吸收光谱分析、核磁共振分析、气相色谱分析等醛类化合物的检测方法[23-27],但它们受到低选择性、基质效应或者复杂的前处理等多方面的限制。基于柱前荧光衍生的高效液相色谱分析法是目前理想的醛类化合物检测手段,荧光衍生可以增强被分析物的疏水性和光谱响应。解决了大部分醛类化合物由于缺少发色团或荧光团带来的检测上的困难。
香豆素结构广泛存在于自然界的许多植物中[28-29],因其具有体积小、荧光量子产率高、斯托克斯位移大,容易合成与修饰等优点,常被应用于荧光探针的设计与合成中[30-40]。含香豆素荧光团的衍生试剂已经在醛类化合物的定量分析中得到了广泛的应用[34-40]。荧光衍生试剂除了需要具备良好的荧光团以外,其分子结构中还必须具备可以在温和条件下快速与被分析物质发生化学反应的基团。由于羟胺能与醛类物质进行硝酮化或者肟化反应的特征,含羟胺结构的化合物也被应用于常见醛类物质和糖类物质的检测中[38-40]。羟胺结构在有机化学中十分重要,被应用于许多经典的有机反应之中[41-44]。传统的N-取代羟胺化合物的合成方法仍是依靠拉西合成,电解还原,肟化水解等传统工艺[38,45-47]。这些方法普遍存在能耗大,所需设备多,工艺流程长,反应条件苛刻,环境污染严重等问题,因而复杂的羟胺化合物难以得到广泛的合成与应用。Rinske等在2013年提出了一种新的合成方案[48],将卤代烃与N,O-双叔丁氧羰基(Boc)羟胺进行反应,得到的产物再通过盐酸水解,就可以得到N-羟胺盐酸盐。该种方法条件温和,原料易得,副产物较少,适合于设计合成较为复杂的N-羟胺衍生物。
Taran课题组发现[49],悉尼酮(又称斯德酮)这种特殊的介离子化合物可以与炔烃进行点击反应,形成带有吡唑环结构的化合物。该反应相比于传统的Husigen环加成反应具有许多明显的优势:可以避免使用较为危险的叠氮化合物,同时悉尼酮的反应活性更高,反应速率更快。除此之外,原本的香豆素-悉尼酮衍生物几乎没有荧光,但与炔烃进行点击反应之后得到的吡唑香豆素产物的荧光性能优异[49]。因此,该类反应近年来多应用于细胞成像等领域[49-52]。
以这些工作为基础,本课题组设计并合成了两种含吡唑香豆素结构的羟胺类荧光探针10和11(Scheme 1和Scheme 2):先根据文献中的方法[52],以4-甲氧基水杨醛和N-乙酰甘氨酸为原料,合成了3-氨基-7-甲氧基香豆素3,3与溴乙酸进行取代反应,得到香豆素羧酸中间体4,4与亚硝酸叔丁酯和三氟乙酸酐反应,生成香豆素-悉尼酮结构的母体化合物5。含炔基的化合物6和化合物7由溴丙炔和含Boc结构的羟胺反应制备[48],再与悉尼酮进行点击反应,得到含有香豆素吡唑结构的中间产物化合物8和化合物9,最后用盐酸脱去Boc保护基生成目标产物10和11。相关的应用研究以N-取代羟胺为例,与糠醛、5-甲基糠醛和5-羟甲基糠醛进行了荧光衍生,通过高效液相色谱-荧光检测法(HPLC-FLD)以及HR-MS(高分辨质谱)进行表征。
1 实验部分
1.1 仪器与试剂
X-4型显微熔点测定仪;Thermo fisher Q-Exactive型质谱仪;Nicolet iS5型红外光谱仪 (KBr 压片);LS55型荧光分光光度计;Bruker AVANCE III 500 MHz NMR型核磁共振仪(氘代二甲亚砜和氘代氯仿为溶剂,TMS为内标)。
所用试剂均为分析纯或化学纯;实验用水为屈臣氏桶装蒸馏水;衍生分析部分涉及到的溶剂均为色谱纯。

Scheme 1
Scheme 2
1.2 合成
(1) 化合物3的合成
将二环己基碳二亚胺(2.48 g, 12.0 mM)和N-乙酰甘氨酸(2.81 g, 24.0 mM)加入至二氯甲烷(40 mL)中搅拌过夜。过滤掉生成的固体并用二氯甲烷(30 mL)冲洗滤渣。合并滤液和洗液,旋蒸浓缩得到白色固体,固体溶解于N,N-二甲基甲酰胺(12 mL)中后,加入4-甲氧基水杨醛(913 mg, 6.0 mM)和乙酸钠(738 mg, 9.0 mM)。将得到的混合物在120 ℃下回流1 h。溶液冷却至室温,加水生成大量黄色固体。过滤,滤渣依次用水(30 mL)和冷乙醇(10 mL)洗涤得到7-甲氧基-3-乙酰氨基香豆素。不经进一步纯化,化合物溶解于浓盐酸(20 mL)、乙醇(10 mL)和水(10 mL)的混合物中。回流反应1 h。冷却至室温,加入碳酸氢钠至无气泡产生。过滤,沉淀用水(100 mL)洗涤,真空干燥得黄色固体化合物3859 mg,产率75%, m.p.137~139 ℃;1H NMR (500 MHz, CDCl3)δ: 7.20(d,J=9.4 Hz, 1H), 6.82~6.81(m, 2H), 6.70(s, 1H), 4.05(s, 2H), 3.84(s, 3H);13C NMR(126 MHz,CDCl3)δ: 159.9, 159.2, 150.5, 129.9, 126.0, 114.6, 112.7, 112.4, 100.9, 55.8; IRνν: 1705, 1646, 1622, 1591, 1506, 1440, 1304, 1272, 1253 cm-1; MSm/z: calcd for C10H10NO3{[M+H]+}192.0661, found 192.0649。
(2) 化合物4的合成
将化合物3(180 mg, 0.94 mM)和溴乙酸(130 mg, 0.94 mM)加入至水10 mL中,在95 ℃下反应3 h。冷却至室温过滤,用水和二氯甲烷洗涤滤渣,干燥后得到化合物495 mg,淡棕色固体,产率41%, m.p.188~190 ℃;1H NMR(500 MHz, DMSO-d6)δ: 7.32(d,J=9.3 Hz, 1H), 6.83~6.86(m, 2H), 6.48(s, 1H), 3.93(s, 2H), 3.82(s, 3H);13C NMR(126 MHz, DMSO-d6)δ: 171.2, 158.7, 157.9, 148.6, 130.5, 126.0, 114.7, 112.4, 106.1, 100.5, 55.6, 44.6; IRν: 3285, 1703, 1633, 1513, 1421, 1274, 1248, 1196, 1159 cm-1; MSm/z: calcd for C12H10NO5{[M+H]+}248.0564, found 248.0564。
(3) 化合物5的合成
将亚硝酸叔丁酯(0.03 mL, 0.25 mM)滴加至化合物4(51 mg, 0.2 mM)的四氢呋喃(3 mL)溶液中,并在室温下搅拌1.5 h之后,加入三氟乙酸酐(0.03 mL, 0.25 mM)继续反应1.5 h。反应结束后,用饱和的NaHCO3水溶液10 mL淬灭混合物。有机相用(3×15 mL)的二氯甲烷萃取,合并后用无水Na2SO4干燥,旋蒸浓缩。采用硅胶柱层析的方法进行提纯分离(EA∶CH2Cl2=5∶95)得到化合物535 mg,黄色固体,产率68%, m.p.188~190 ℃;1H NMR(500 MHz, DMSO-d6)δ: 8.45(s, 1H), 7.61(d,J=8.8 Hz, 1H), 7.32(s, 1H), 7.03(dd,J=8.7 Hz, 1.5 Hz, 1H), 6.94(d,J=2.3 Hz, 1H), 3.96(s, 3H);13C NMR(126 MHz, DMSO-d6)δ: 168.8, 166.0, 156.4, 154.5, 138.2, 131.2, 116.8, 115.2, 110.4, 101.1, 97.4, 56.4; IRν: 3285, 1703, 1633, 1513, 1421, 1274, 1248, 1196, 1159 cm-1; MSm/z: calcd for C12H9N2O5{[M+H]+}261.0506, found 261.0504。
(4) 化合物6的合成
将溴丙炔(0.980 mg, 7.36 mM)缓慢滴入至溶解有N-羟基氨基甲酸叔丁酯(0.967 g, 7.72 mM)和碳酸钠(1.560 g, 14.7 mM)的DMF(N,N-二甲基甲酰胺)溶液中,加热至70 ℃搅拌过夜。反应结束后混合物加入适量水,用乙酸乙酯萃取三次。有机相用饱和氯化钠溶液(50 mL)洗涤,无水硫酸钠干燥后真空浓缩。粗产物经硅胶柱层析纯化 (PE∶EA=10∶1)得化合物6510 mg,无色油状液体, 产率41%;1H NMR(500 MHz, CDCl3)δ: 4.34(br.s, 2H), 2.36(t, 1H), 1.34(s, 9H);13C NMR(126 MHz, CDCl3)δ: 156.4, 82.1, 78.2, 75.6, 63.7, 28.2(3C); IR(KBr)ν: 3291, 2980, 1725, 1478, 1393, 1369, 1251, 1166 cm-1; MSm/z: calcd for C8H14NO3{[M+H]+}172.0968, found 172.0964。
(5) 化合物7的合成
将溴丙炔(80%甲苯溶液, 0.23 mL, 2.07 mM)滴加至N,O-二Boc-羟胺(0.435 g, 1.86 mM)和K2CO3(0.343 g, 2.48 mM)的DMF(15mL)溶液中。混合物常温搅拌反应16 h后加入水(100 mL),乙酸乙酯(2×50 mL)萃取混合物。有机相合并后依次用水(2×100 mL)和饱和食盐水(100mL)洗涤,用无水硫酸钠干燥。在旋转蒸发仪上除去溶剂,得到化合物70.304 g为无色油状液体,产率60%;1H NMR(500 MHz, CDCl3)δ: 4.31(br.s, 2H), 2.26(t, 1H), 1.51(s, 9H), 1.48(s, 9H);13C NMR(126 MHz, CDCl3)δ: 154.5, 151.9, 84.9, 83.3, 76.9, 72.6, 40.4, 28.0(3C), 27.5(3C); IR (KBr)ν: 3293, 2982, 1787, 1726, 1477, 1371, 1280, 1232 cm-1; MSm/z: calcd for C13H22NO5{[M+H]+} 272.1498, found 272.1500。
(6) 化合物10的合成
将化合物5(200 mg, 0.812 mM)与化合物6(166 mg, 0.896 mM)溶解于N,N-二甲基甲酰胺(8 mL)中,再配置含有五水合硫酸铜(40.6 mg, 0.164 mM)、 BPDS(4,7-二苯基-1,10-菲啰啉二磺酸二钠盐水合物, 87.2 mg, 0.164 mM)以及三乙醇胺(121.6 mg, 0.812 mM)的水溶液,并加入至上述的DMF溶液中,最后再加入抗坏血酸钠(321.7 mg, 1.824 mM),50 ℃下搅拌反应40 min。反应结束后加入0.05 M HEDTA水溶液(50 mL),用乙酸乙酯萃取3次。有机相用饱和氯化钠溶液(50 mL)洗涤,无水硫酸钠干燥后真空浓缩,粗产物经硅胶柱层析纯化(PE∶EA=3∶1)得131 mg白色粉末8。取100 mg化合物8溶解于盐酸-乙酸乙酯混合溶液6 mL中(V∶V=1∶5), 30 ℃下反应,用TLC监测反应进程,待原料消失后停止反应,在旋转蒸发仪上除去溶剂,得化合物1065 mg为白色粉末,产率78%, m.p. 186~188 ℃;1H NMR(500 MHz, DMSO-d6)δ: 10.99(s, 3H), 8.53(s, 1H), 8.50(s, 1H), 7.90(s, 1H), 7.83(dd,J=8.5 Hz, 1.5 Hz, 1H), 7.13(s, 1H), 7.05(dd,J=8.5 Hz, 1.5 Hz, 1H), 5.04(s, 2H), 3.89(s, 3H);13C NMR(126 MHz, CDCl3)δ: 163.0, 156.8, 154.0, 142.2, 132.5, 130.5, 123.0, 115.8, 113.8, 112.3, 100.9, 87.0, 56.5; IRν: 2989, 2975, 1689, 1622, 1514, 1445, 1399, 1045 cm-1; MSm/z: calcd for C14H14N3O4{[M]+}288.0984, found 288.1016。
(7) 化合物11的合成
将化合物5(200 mg, 0.812 mM)与化合物7(280 mg, 0.896 mM)溶解于N,N-二甲基甲酰胺(8 mL)中,再配制含有五水合硫酸铜(40.6 mg, 0.164 mM)、 BPDS(4,7-二苯基-1,10-菲啰啉二磺酸二钠盐水合物87.2 mg, 0.164 mM)以及三乙醇胺(121.6 mg, 0.812 mM)的水溶液,并加入至上述的DMF溶液中,最后再加入抗坏血酸钠(321.7 mg, 1.824 mM),50 ℃下搅拌反应40 min。反应结束后加入0.05 M HEDTA水溶液(50 mL),用乙酸乙酯萃取三次。有机相用饱和氯化钠溶液(50 mL)洗涤,无水硫酸钠干燥后真空浓缩,粗产物经硅胶柱层析纯化(PE:EA=3:1)得180 mg淡黄色液体9。取100 mg化合物9溶解于盐酸-乙酸乙酯混合溶液6 mL中(V:V=1:5),于30 ℃反应至终点,用TLC监测反应进程。待原料消失后停止反应,在旋转蒸发仪上除去溶剂,得化合物1152 mg为白色粉末,产率82%, m.p.215~216 ℃;1H NMR(500 MHz, DMSO-d6)δ: 11.59(br. s, 1H), 10.99(br. s, 1H), 8.57(s, 1H), 8.51(s, 1H), 7.90(s, 1H), 7.83(dd,J=8.5 Hz, 1.5 Hz, 1H), 7.12(s, 1H), 7.05(dd,J=8.5 Hz, 1.5 Hz, 1H), 4.32(s, 2H), 3.89(s, 3H);13C NMR(126 MHz, CDCl3)δ: 163.0, 156.8, 154.0, 142.8, 133.0, 132.2, 130.5, 123.0, 113.8, 112.3, 111.7, 100.9, 56.5, 44.8; IRν: 2971, 2901, 1717, 1605, 1508, 1442, 1402, 1066 cm-1; MSm/z: calcd. for C14H14N3O4{[M]+}288.0984, found 288.1018。
1.3 探针的荧光量子产率测定
根据文献报道[46]及实验结果显示,香豆素-悉尼酮的原母体结构几乎不产生荧光, 但在与炔基化合物进行点击反应以后,其荧光性能会得到大大增强,荧光量子产率有明显提高。采用1 μM硫酸奎宁(溶于0.1 M硫酸溶液)作为标准参照物,将1 mM 羟胺溶液用水溶液稀释成1 μM,然后对其进行相关检测。
1.4 探针分子对糠醛的定量衍生分析
工业上糠醛检测所采用的国标法即为盐酸羟胺肟化法[55],但是该方法误差大,配制标准溶液的耗时较长,因而存在很大的局限性。为此,根据合成的羟胺荧光探针的特性,以N-取代羟胺探针11为例,设计了新的验证探针分子与糠醛定量衍生的方法,详细步骤为:在1.5 mL EP管中依次加入40 μL 探针溶液(0.2 mM,去离子水溶解)、20 μL醛的标准溶液(0.2 mM,纯甲醇溶解)和20 μL 2-氨基-5-甲氧基苯甲酸(40 mM,纯甲醇溶解),最后加入20 μL甲醇,混匀后置于20 ℃下反应40 min。反应后的混合物取10 μL直接注射进色谱系统中,高效液相色谱采用梯度洗脱模式,洗脱程序如表1所示,其中流动相A为乙腈-水(0.2/0.8,V/V),流动相B为乙腈-水(0.5/0.5,V/V)。A、B相中均加入0.02 M的乙酸和0.01 M的乙酸钠,流速为1 mL/min。荧光检测器激发波长设为340 nm,发射波长设为407 nm,柱温箱为常温。

表1 梯度洗脱的时间程序
1.5 方法学验证试验
荧光定量分析的回归方程通过以下方法获得:选取浓度范围从定量限(LOQ)到4 μM的糠醛样品用于荧光衍生,然后线性拟合荧光衍生物所对应的色谱峰的面积和相应的浓度得到回归方程和对应的相关系数。
2 结果与讨论
1.2 合成
本文合成的吡唑香豆素结构的荧光探针,与只含有香豆素结构的荧光探针相比,不仅有更优异的荧光性能,同时合成的香豆素-悉尼酮荧光砌块(化合物5)可以与多种炔基化合物发生点击反应,更利于与多种不同的反应基团进行连接,合成出一系列检测不同小分子的含吡唑香豆素结构的荧光探针。
λ/nm
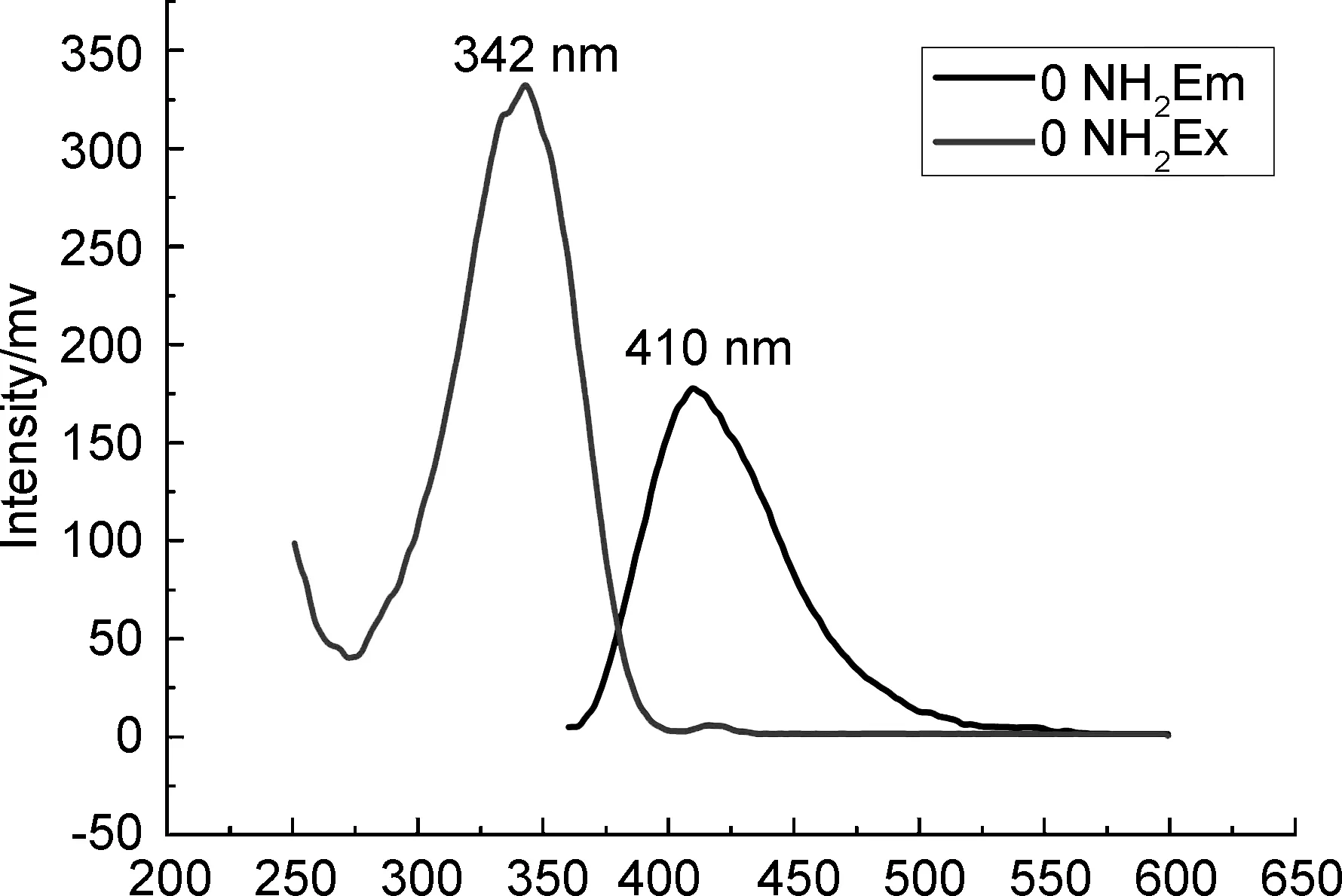
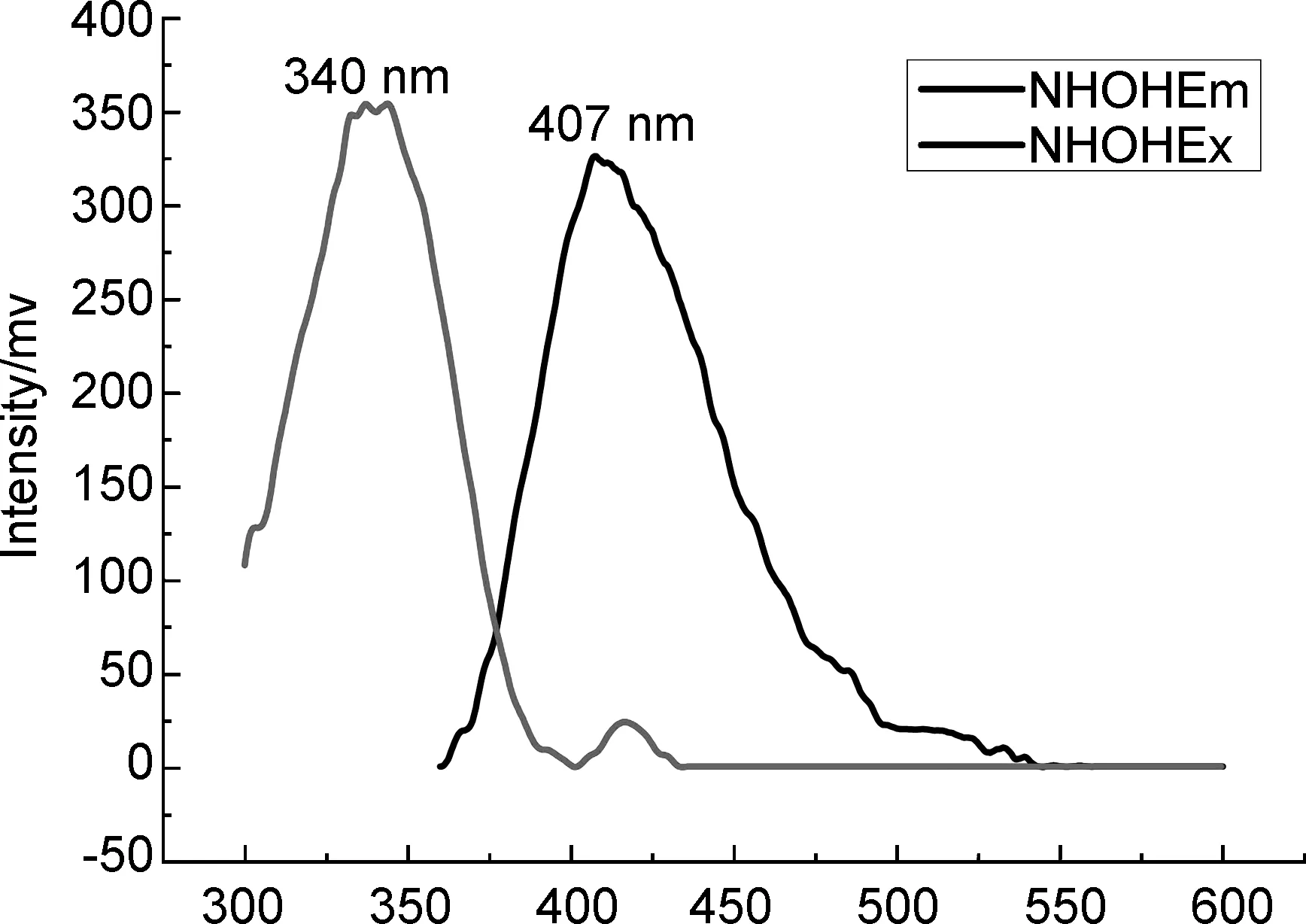
λ/nm图1 化合物10和11的荧光谱图

Figure 1FL spectra of 10 and 11
最初的实验方案是直接将含有两种羟胺结构的端炔与悉尼酮-香豆素进行点击反应得到所需要的荧光探针,但是羟胺存在极性较大、活性较高的问题,对点击反应产生了干扰。于是本实验采用引入保护基的方案来减少点击反应中副反应的发生,在经过多次尝试后,最终选择并合成了含有Boc基团的炔基化合物6和7,以便于点击反应能够顺利进行,同时提高产率,减少副产物的产生。
N-取代羟胺合成的传统方法一般通过醛类化合物发生肟化反应得到含有肟式结构的中间体,然后与强还原剂发生还原反应得到相应的羟胺化合物(Scheme 3)。该类反应往往条件复杂,副产物较多,不利于较为复杂的羟胺化合物的合成。而本文采用的方案较为简便,条件温和,有望用于其他N-取代羟胺化合物的合成之中。
2.2 10与11的荧光光谱及荧光量子产率
化合物10与11的荧光光谱见图1。由图1可知,两种化合物的斯托克斯位移分别为67 nm和68 nm,相应的物理常数见表2。综合图1和表2可知,新合成的含吡唑香豆素结构的探针具有良好的荧光量子产率。
Time/min
2.3 化合物11与糠醛的衍生分析结果
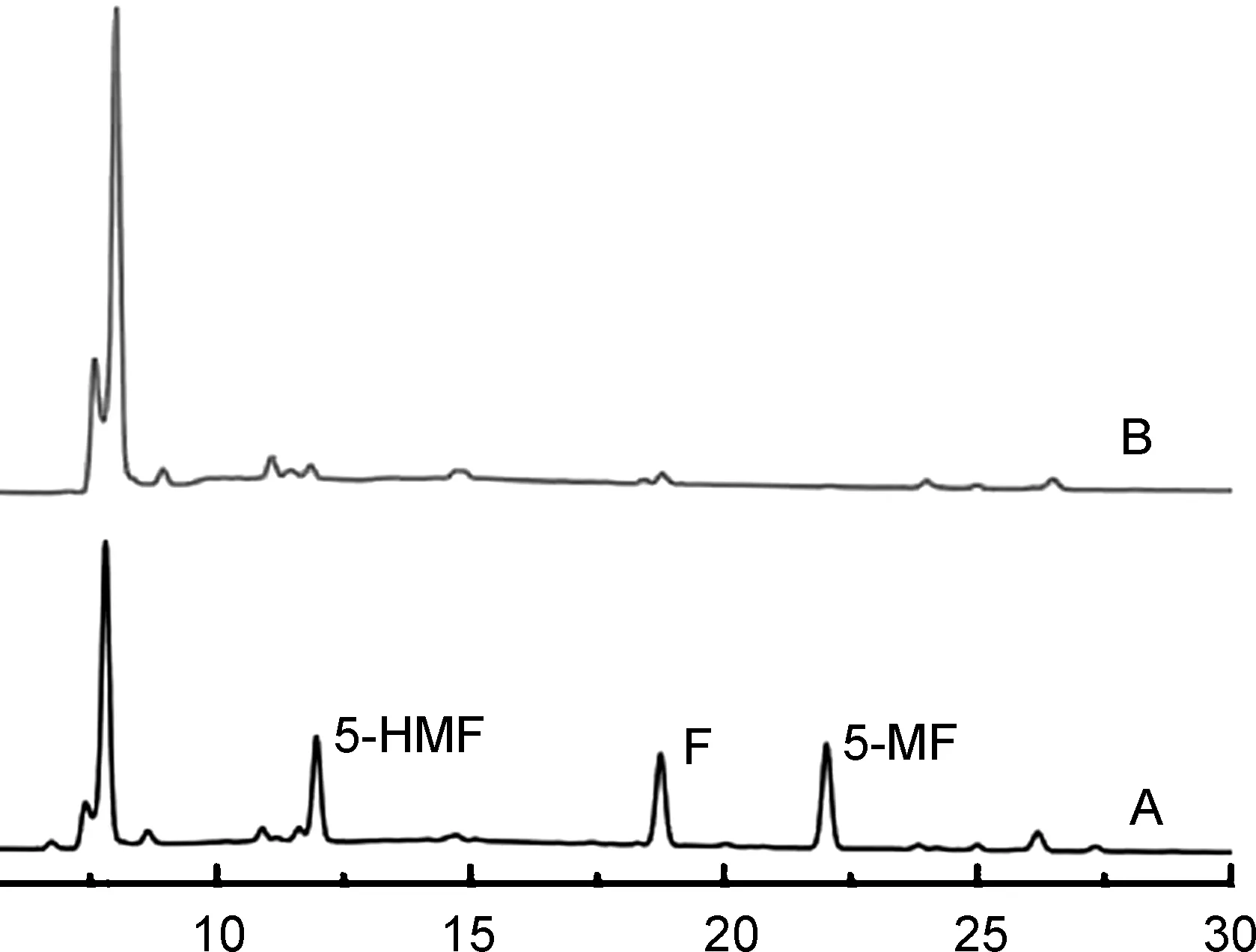
化合物11与糠醛的衍生结果如图2所示。相较于空白样,在加入混合糠醛以后,色谱图中分别在12 min、 18 min、 22 min出现了3个新峰,对应的是3种糠醛的具有硝酮结构的衍生产物(N-取代羟胺基香豆素与呋喃醛的硝酮化反应)。这与其他研究报道的结果类似[38-40,56-57]。而其他常用的检测试剂,例如2,4-二硝基苯肼(DNPH)和邻-(2,3,4,5,6-五氟苄基)羟胺盐酸盐(PFBHA)在与糠醛类物质反应时,在色谱图上会有两个主要的衍生产物峰。根据肟化反应原理,这两种衍生试剂在与羰基类物质反应时会生成顺-反两种异构体[38-40,58-59],而本文设计的方法只生成单一的衍生产物峰,且具有较高的灵敏性,因此可用于准确测定糠醛类物质。
随后,继续通过质谱分析,进一步证实了衍生产物的生成。质谱图(图略)中的衍生物质谱峰信号强烈,表明吡唑环结构中两个氮原子的存在有助于质谱的离子化。因此,本文合成的香豆素吡唑羟胺荧光探针也有望在单独基于质谱分析的醛类物质检测中发挥重要作用。

表3 硝酮化反应分析糠醛混合物所得线性范围、线性方程、检出限和相对标准偏差
2.2 方法学验证
本分析方法的线性范围、回归方程、相关系数(R2)和检出限如表3所示。由表3可知,以3种糠醛定量限(LOQ, S/N=10)的浓度到 4000 nM 为线性范围,在此范围内具有较好的线性关系,相关系数R2为0.9997~0.9999;检测限(LOD, S/N=3)为0.4~0.8 nM,能够满足食品中糠醛类化合物含量的检测。
以香豆素-悉尼酮为荧光合成砌块,通过它与Boc基团保护的炔基羟胺的点击化学反应最终得到了两种具有吡唑香豆素结构的羟胺类荧光探针。由于吡唑香豆素结构的存在,其荧光性能良好,N-取代和O-取代探针的荧光量子产率分别达到0.72和0.76。之后以N-取代羟胺为例,分别与食品中常见的糠醛、5-甲基糠醛和5-羟甲基糠醛进行了荧光衍生,并用HPLC-FLD以及HR-MS进行了分析表征。初步的研究结果表明:三种糠醛衍生后形成的硝酮化合物在HPLC-FLD中实现了基线分离,具有较强的荧光衍生峰和色谱学行为,当三种糠醛的浓度在0~4000 nM时,荧光强度与浓度有着良好的线性关系,相关系数R2良好(≥ 0.9997),检测限较低(≤ 0.8 nM),重现性好。吡唑环结构中两个氮原子的存在,有助于衍生产物在质谱分析中的离子化,并增强质谱峰的信号强度。
