喹唑啉类端锚聚合酶抑制剂3D-QSAR和分子对接研究
2022-03-23陈小中林治华
陈小中,沈 燕,王 娟,胡 勇,林治华
(重庆理工大学 药学与生物工程学院, 重庆 400054)
端锚聚合酶(TNKS)是聚腺苷二磷酸核糖聚合酶(PARP)蛋白家族成员之一[1]。TNKS参与多种生物学进程,如有丝分裂、Wnt信号通路等[2]。经研究发现,Wnt信号在多种癌症中被过度激活,抑制TNKS能稳定Wnt信号转导,从而抑制肿瘤细胞增殖[3]。因此,TNKS已成为肿瘤治疗领域中一个具有吸引力的靶点[4]。在部分端锚聚合酶抑制剂研究方面,诺华研究人员在2009年报道了小分子化合物XAV939(IC50=11 nmol),其通过结合TNKS烟酰胺结合位点从而抑制TNKS活性[5]。Texas大学研究人员通过基于细胞评价的方法,从220 000个化合物中筛选出TNKS抑制剂endo-IWR-1,通过结合TNKS烟酰胺结合位点从而抑制TNKS活性。默克公司研究人员设计合成了一系列TNKS抑制剂,在这些化合物中,化合物5K在细胞实验(IC50=7 nmol)和肿瘤移植模型小鼠中皆显示良好的抗肿瘤作用。如图1所示,列举了具有代表性的TNKS抑制剂结构。
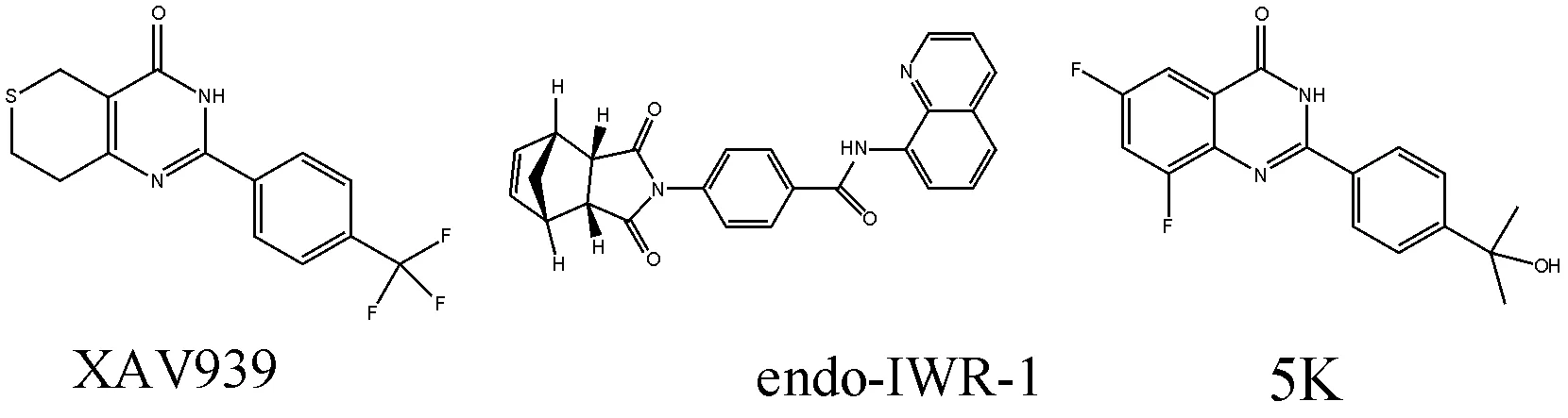
图1 代表性的TNKS抑制剂结构
目前,尚未有TNKS抑制剂被批准应用于临床,但已有PARP家族其他成员的抑制剂(如olaparib)应用于临床。针对TNKS的抑制剂正在开发中,截至目前,尚未有专门的选择性TNKS抑制剂上市。TNKS显示了巨大的临床应用开发潜力,是具有吸引力的抗癌药物靶标之一。
3D-QSAR研究包括分子力场分析法(CoMFA)[6]和比较分子相似性法(CoMSIA)[7]。CoMFA和CoMSIA是药物设计领域中研究一系列化合物定量构效关系的2种经典的方法[8]。分子对接是一种表征受体与其配体结合方式的方法[9]。3D-QSAR和分子对接可用于研究化合物的详细药效团特征,同时为设计新化合物提供有用信息[10]。有研究表明,3D-QSAR和分子对接的联合应用能够发现新的先导化合物。目前尚无喹唑啉衍生物作为TNKS抑制剂的3D-QSAR研究报告。因此,在本文中,利用CoMFA、CoMSIA和分子对接技术研究一系列喹唑啉类TNKS抑制剂。该研究可为新型TNKS抑制剂的开发提供理论参考。
1 实验方法
1.1 数据集
数据集来源于Buchstaller等[1]报道的28个喹唑啉类TNKS抑制剂。所有抑制剂均使用SYBYL.2.1软件构建。抑制剂的结构及其pIC50(-logIC50)值如表1所示。适当的能量最小化分子方法对获得准确的3D-QSAR模型至关重要。使用Tripos力场[11]和Powell方法使化合物能量最小化,最大迭代次数设置为1 000,其他参数取默认值。所有化合物加载Gasteiger-Hückel电荷[12]。训练集和测试集分别包含22种和6种化合物, 测试集用*标识,如表1所示。所有化合物使用喹唑啉骨架进行叠合(图2)。
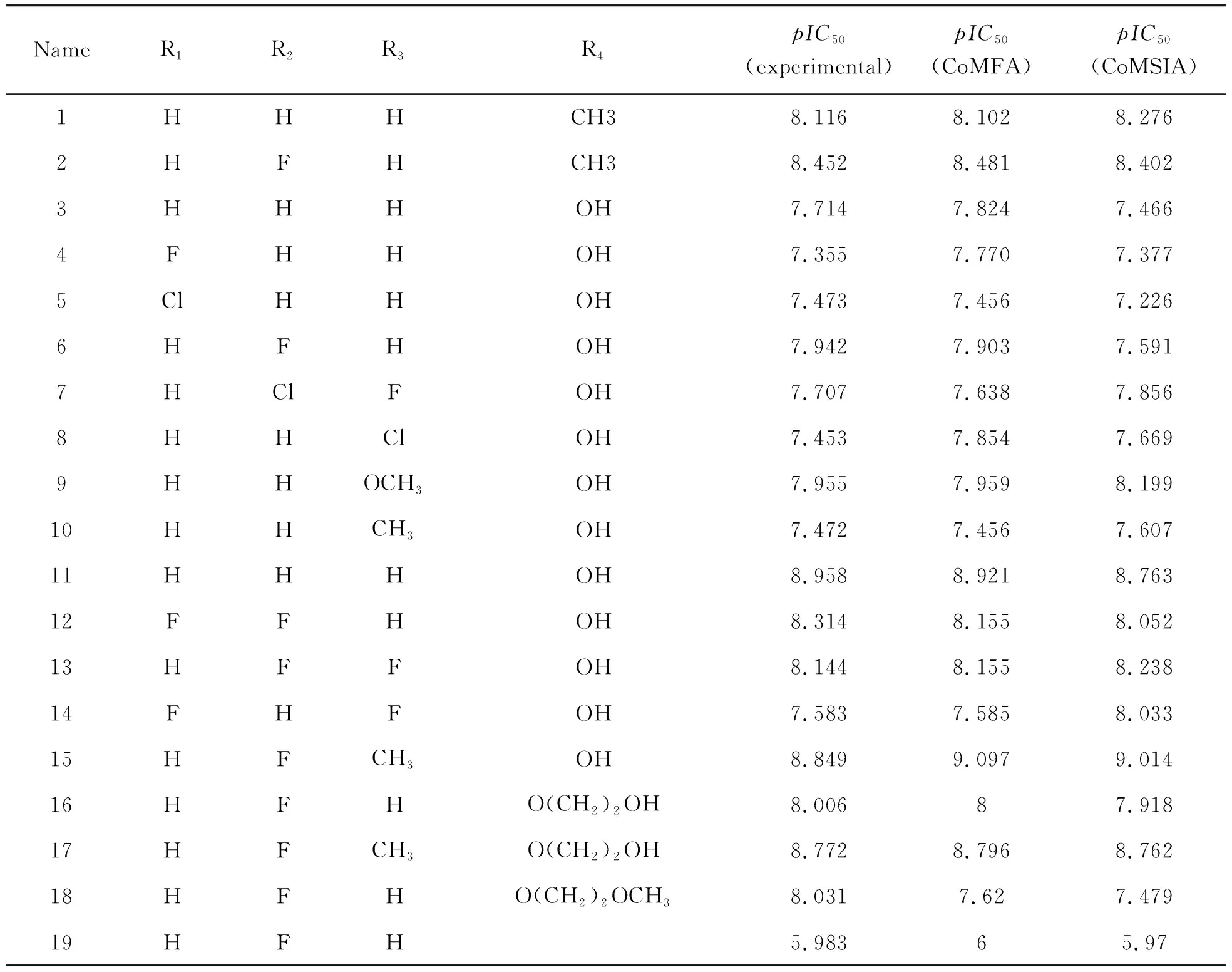
表1 TNKS抑制剂结构及CoMFA和CoMSIA模型的预测值
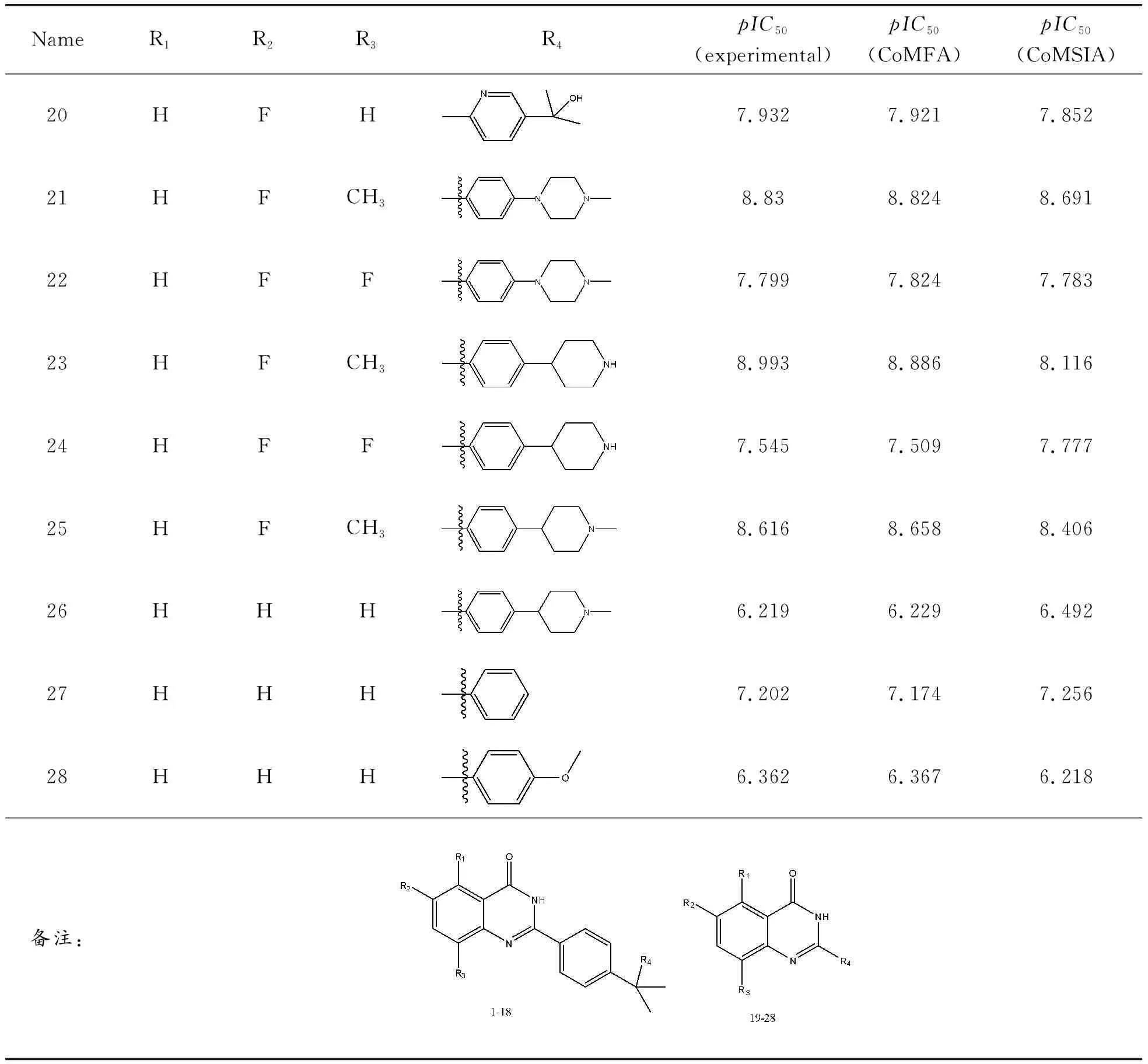
续表(表1)

图2 28个TNKS抑制剂基于公共骨架的叠合图
1.2 建立CoMFA CoMSIA模型

(1)
(2)
(3)

1.3 分子对接
分子对接(molecular docking)依据“锁-钥原理”研究受体生物大分子和配体小分子之间的相互作用,利用特定算法计算“钥匙”(配体小分子)与“锁”(受体生物大分子)在几何形状、静电相互作用、疏水作用等方面的互补匹配情况。Suflex具有较高的准确度,故本研究使用SYBYL-X2.1软件的Suflex模块进行对接研究[13]。从Protein Data Bank(https://www.rcsb.org)下载TNKS与化合物13复合物的x射线晶体结构,PDB代码为6qxu。Suflex具有3种主要的对接模式,包括Screen(高通量筛选模式)、Geom(标准模式)和GeomX(高精度模式)。为了获得准确的对接结果,使用GeomX模式进行对接。首先,进行蛋白准备,提取出PDB文件中的原始配体,并删除其中的水分子;其次,给蛋白质加上氢键,蛋白质活性口袋在原始配体的基础上生成;最后,将配体对接至生成的TNKS活性口袋中。Suflex使用联合打分函数,将得分最高的构象作为最后的分子对接结果。
2 结果与讨论
2.1 CoMFA和CoMSIA模型相关参数
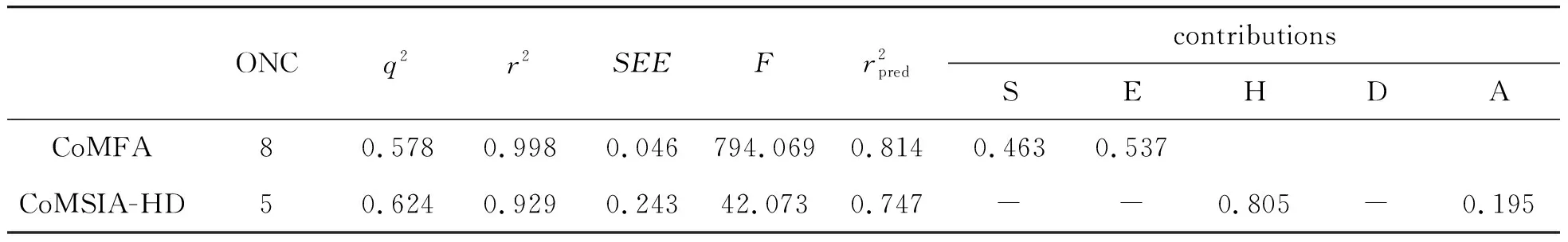
表2 CoMFA和CoMSIA分析的统计参数
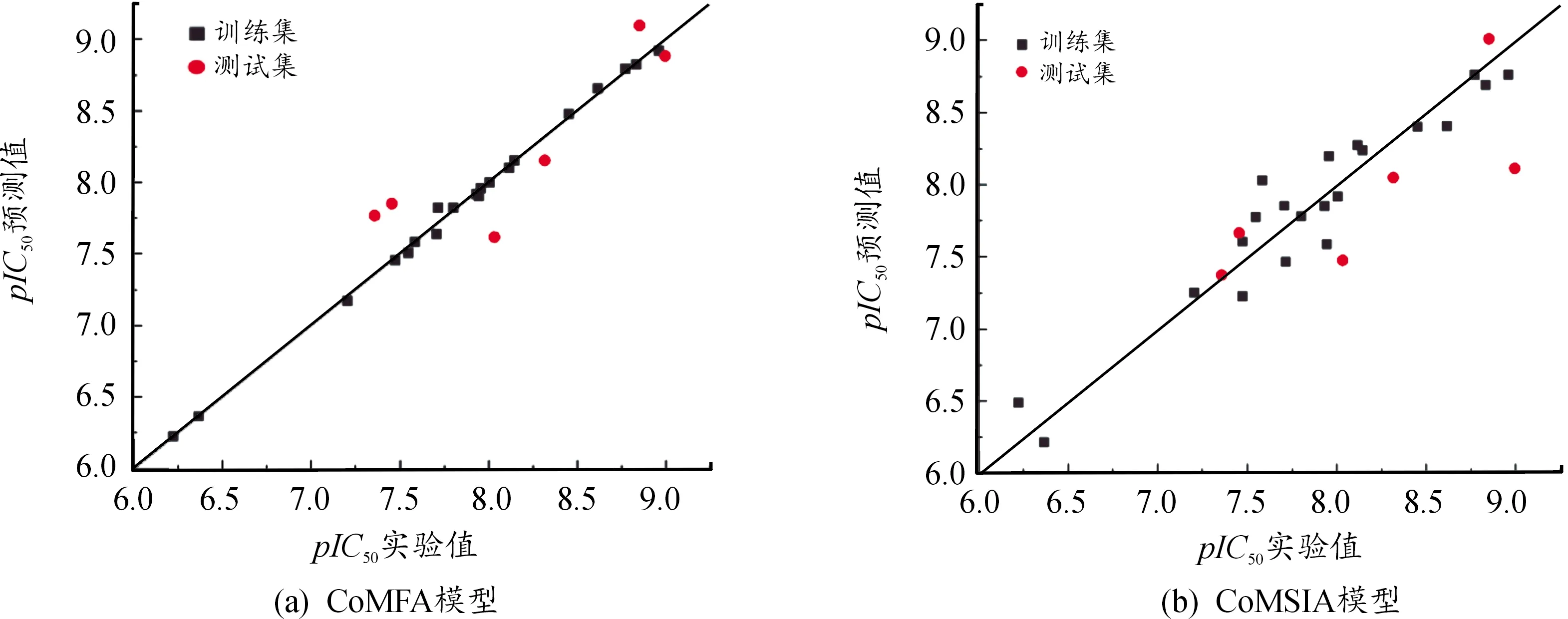
图3 实验pIC50值与预测pIC50值散点图
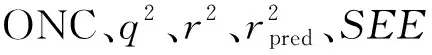
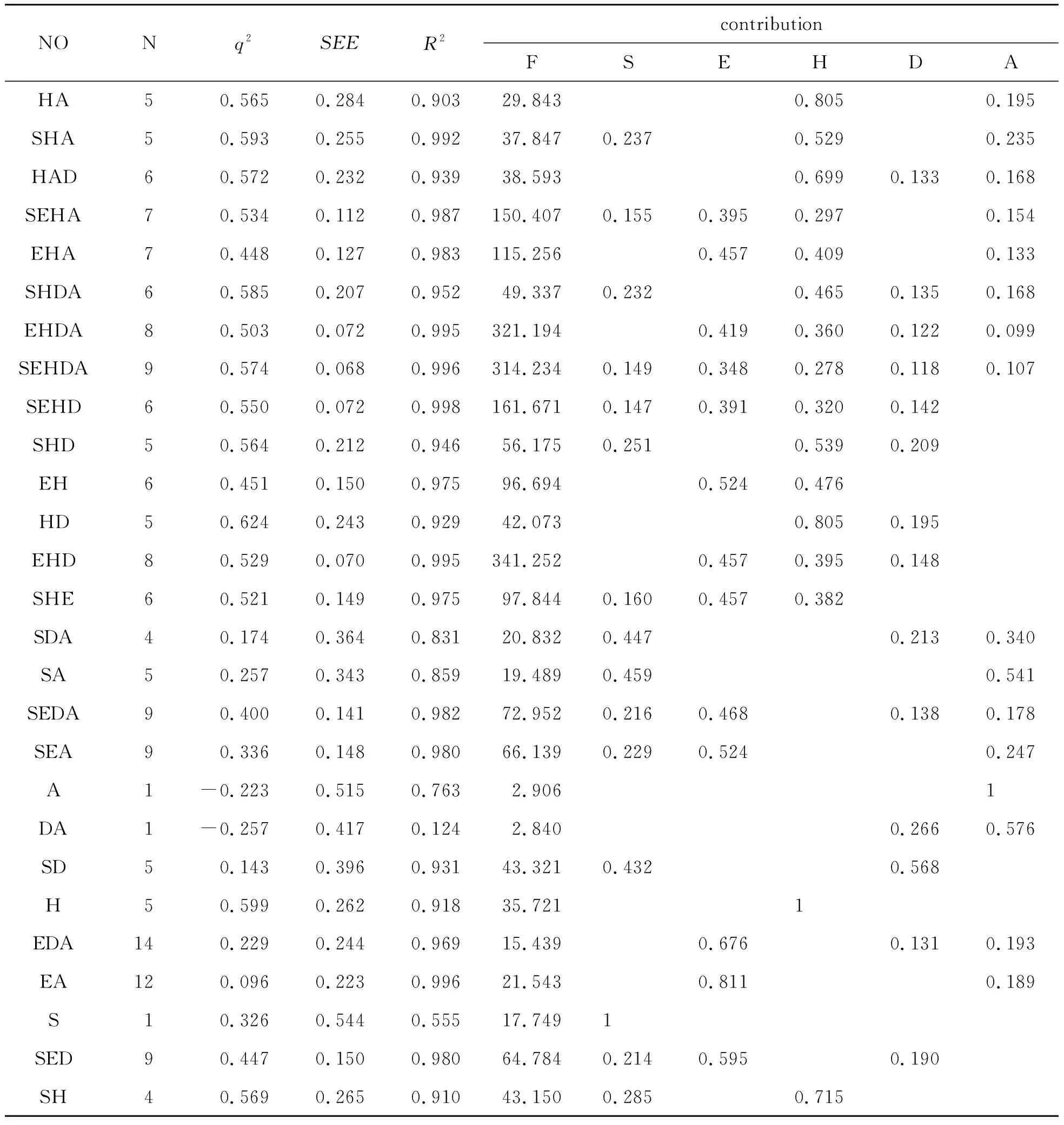
表3 33种可能的CoMSIA模型的偏最小二乘(PLS)结果
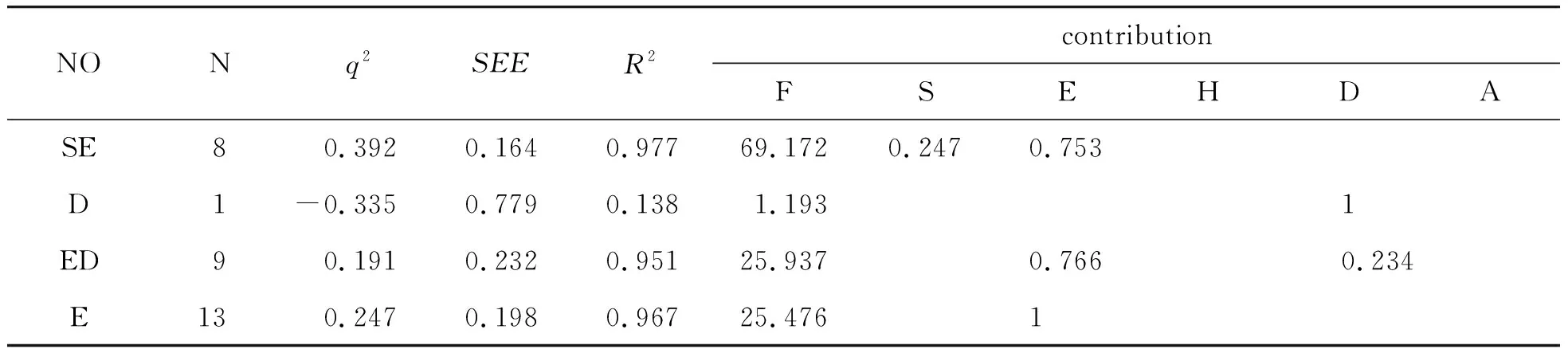
续表(表3)
2.2 CoMFA等势图
CoMFA等势图如图4所示,化合物11在图中以供参考。蓝色区域(贡献度为80%)表示吸电子基团对活性有利的区域,红色区域(贡献度为20%)表示吸电子基团对活性不利的区域。在R4位置的芳香环附近有一个较大的蓝色色块,表明此区域存在吸电子基团,将会增加化合物活性。这也许是化合物8(pIC50=7.95)的活性高于化合物9(pIC50=7.85)的原因。绿色区域(贡献度为80%)表示空间位阻大的基团对活性有利,黄色区域(贡献度为20%)表示空间位阻大的基团对活性不利。在R3上方有一个绿色色块,表明此处空间位阻大的取代基会增加活性。这可以解释为什么化合物11(R3=CH3)的活性比化合物9(R3=F)的活性高。
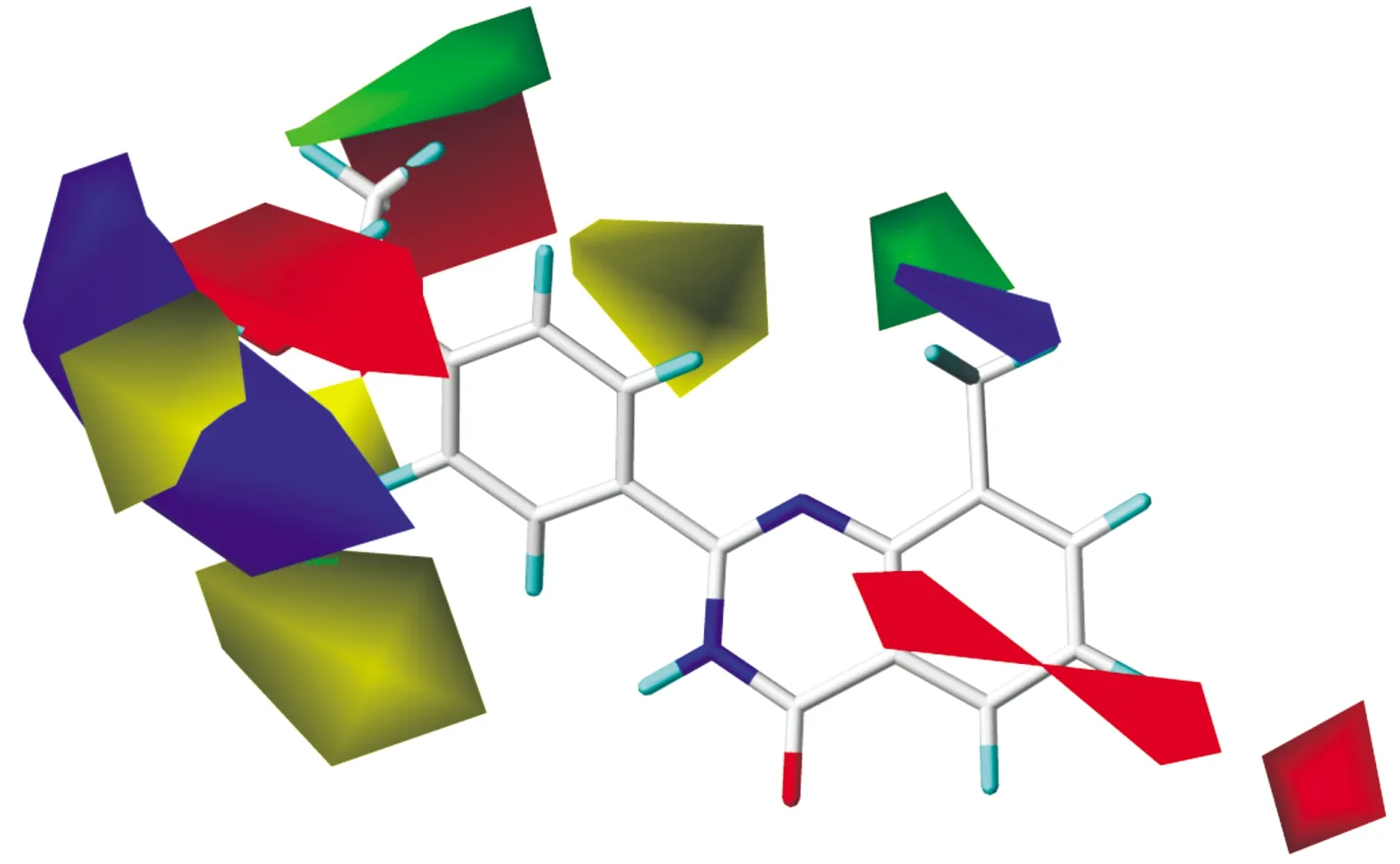
图4 CoMFA等势图
2.3 COMSIA等势图
CoMSIA等势图如图5所示,化合物15在图中以供参考。黄色区域(贡献度80%)和白色区域(贡献度为20%)分别表示了具有疏水性和亲水性基团对活性有利的区域。R3位置有一个黄色色带,这可以解释在苯环的R3位置具有疏水基团(-CH3)的化合物1比化合物30活性(R3=H)高的原因。青色部分表明此区域氢键供体基团(贡献度为80%)会提高化合物活性。相反地,紫色部分(占20%的贡献)表示氢键供体基团在该区域会降低化合物活性。2个青色色块位于R3芳基附近,表明该部分上的氢键供体基团将增加化合物活性。例如,R3位置芳环上有哌啶基的化合物23(pIC50=8.88)的活性比R3位置具有哌嗪基的化合物22(pIC50=7.82)的活性高。
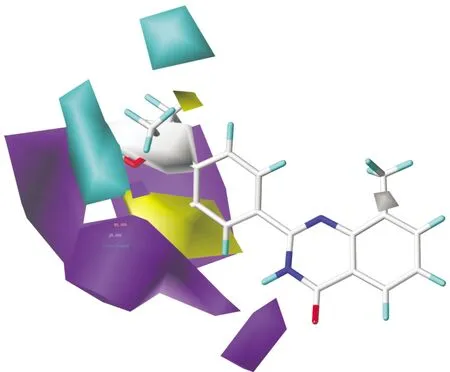
图5 CoMSIA等势图
3 分子对接
为了更好地解释TNKS及其抑制剂之间的结合方式,对活性最高的化合物15和活性最低的化合物19分别与TNKS进行分子对接。化合物15和19被对接在TNKS烟酰胺结合位点,这2个化合物与TNKS之间的二维相互作用如图6所示(图片由Discovery Stido 2018生成的)。化合物15与TNKS氨基酸残基Ser1221、Gly1185发生氢键相互作用,这与化合物19相同。具体来说,即化合物15和19吡唑环上的羰基氧原子和-NH氢原子分别作为氢键与Gly1185形成2个氢键。化合物11与TNKS复合物x射线晶体结构(PDB ID:6qxu)显示,化合物11和TNKS残基Ser1221、Gly1185形成与化合物15和19相同的3个氢键,表明Ser1221、Gly1185在稳定TNKS过程中有重要作用。值得注意的是,CoMSIA等势图表明喹唑啉附近存在紫色色块,表明该区域可能与TNKS发生氢键相互作用,分子对接结果和等势图结果相符。化合物15和19吡唑环与残基Tyr1213、Tyr1224产生π-π堆积作用。化合物15和化合物19的氟原子(R2)与Phe1214发生卤素相互作用。化合物15的甲基与TNKS残基Tyr1203、Tyr1213、Tyr1224和Lys1220产生π-Alkyl作用。化合物19的R3位置无甲基,所以没有上述类似作用。

图6 TNKS与化合物15、化合物19的分子对接相互作用示意图
4 结论
采用经典的定量构效关系分析方法CoMFA和CoMSIA对一系列喹唑啉类似物进行研究,发现3D-QSAR模型具有良好的统计参数,证明了这些抑制剂的结构与活性之间的相互关系。分子对接研究结果揭示了这些抑制剂与TNKS之间相互作用的模式,证实了CoMFA和CoMSIA模型。该研究是计算机辅助药物方法在TNKS方面的应用,可为开发TNKS抑制剂作为抗癌药物提供参考。
