Ti掺杂对Li-Mg-N-H材料储氢性能影响的第一性原理研究
2022-03-19闫敏艳宫长伟张敏刚
闫敏艳,宫长伟,张 鹤,张敏刚
(太原科技大学材料科学与工程学院,太原 030024)
0 引 言
发展氢能是解决人类当前所面临的能源危机和环境问题的一种重要途径,目前制约其规模化应用的瓶颈主要在于高效、安全、经济的储氢技术。近十多年来,金属氮氢化物、铝氢化物、硼氢化物等多种轻质高容量储氢材料的迅猛发展为储氢技术的突破带来希望[1]。其中,Li-Mg-N-H材料因具有较高的可逆储氢容量(约5.6%,质量分数)和适中的热力学性能,是一种具有良好应用前景的轻质储氢材料。但是,较慢的放氢动力学和较高的工作温度极大地限制了其应用[2]。
Li-Mg-N-H材料是由LiNH2/MgH2或Mg(NH2)2/LiH组成的复合储氢体系,其吸放氢反应的动力学势垒主要来自反应物中化学键的断裂和重构所需要克服的能量。掺杂改性是降低反应能垒、提高吸放氢动力学性能的一种有效手段,为改善Li-Mg-N-H材料的储氢性能,人们开展了大量实验研究工作[3-11]。Ma等[8]研究发现,石墨担载的Ru纳米颗粒可促进氨基物和亚氨基物之间的界面反应并加快N—H的断裂与重构,进而有效改善Mg(NH2)2-2LiH材料的吸放氢动力学性能,且经十次吸放氢循环后样品的放氢动力学未见衰减。Zhu等[9]研究发现,TiH2、NbH和VH等掺杂可使LiNH2+MgH2体系的起始放氢温度降低,放氢动力学加快。Shahi等[10-11]研究了VCl3、V2O5和TiO2等掺杂对Mg(NH2)2-2LiH材料储氢性能的影响及其作用机理。推测分析,V和Ti均可通过与Mg(NH2)2中N原子的孤对电子发生作用而形成不稳定中间产物,改变了该材料的放氢反应路径,从而使其放氢起始温度降低,放氢动力学加快。
前人研究表明,过渡金属及其化合物掺杂在Li-Mg-N-H材料储氢性能改善方面效果显著,但相关掺杂改性机理尚不明确,且缺乏理论研究支撑。借助理论计算手段,弄清其具体掺杂改性机理,对于新型高效催化剂的设计开发及Li-Mg-N-H材料的实用化具有重要指导意义。目前,有关Li-Mg-N-H材料的理论研究工作主要体现在运用第一性原理理论计算手段分析并确定Li2MgN2H2的晶体结构[12-13],及对其电子结构、成键特性和贮氢热力学性能进行研究等[14-16]。此外,Wang等[17]通过对比未掺杂Li2MgN2H2和Ti替换(100)面上的Li后形成的Li7TiMg4(N2H2)4的态密度和Li/H空位形成能,分析了Ti掺杂对Li-Mg-N-H材料储氢性能的影响。但是,现有报道中,并未对Ti元素在Li2MgN2H2结构中的稳定替代位置进行研究,且未对Ti掺杂前后Li2MgN2H2材料的生成焓及电子结构进行系统研究以阐明其具体掺杂改性机理。因此,本文将采用基于密度泛函理论的第一性原理赝势平面波方法,对Ti替换Li2MgN2H2中不同位置的Li或Mg的杂质替换能进行系统计算,以确定其稳定替代位置。在此基础上,进一步详细研究Ti掺杂对Li2MgN2H2材料的生成焓、能带结构、态密度、差分电荷密度及布居等的影响情况,以期从电子结构层次上阐明Ti掺杂对Li-Mg-N-H材料吸氢动力学性能的影响机理,为该材料储氢性能的进一步改善提供理论指导。
1 计算模型与方法
1.1 计算模型
Li2MgN2H2材料按照温度从低到高存在α-Li2MgN2H2(室温~330 ℃)、β-Li2MgN2H2(330~500 ℃)和γ-Li2MgN2H2(500 ℃以上)三种不同的相结构,因该材料吸放氢反应温度通常不超过400 ℃,因此,本文选择低温相α-Li2MgN2H2进行研究。α-Li2MgN2H2为正交结构,空间群为Iba2(No.45),晶胞参数为:a=0.978 71 nm,b=0.499 27 nm,c=0.520 19 nm,各原子的具体占位信息如表1所示[12]。该结构中Li/Mg原子在4b和8c位置的占位不明确,Rijssenbeek等[12]和Wang等[16]指出Li/Mg原子的均匀分布有利于形成更为稳定的晶体结构,并通过对Li2MgN2H2可能存在的三种结构的总能进行计算,确定出能量最低、构型最稳定的晶体结构,如图1所示,单个晶胞中包含8个Li、4个Mg、8个N和8个H。
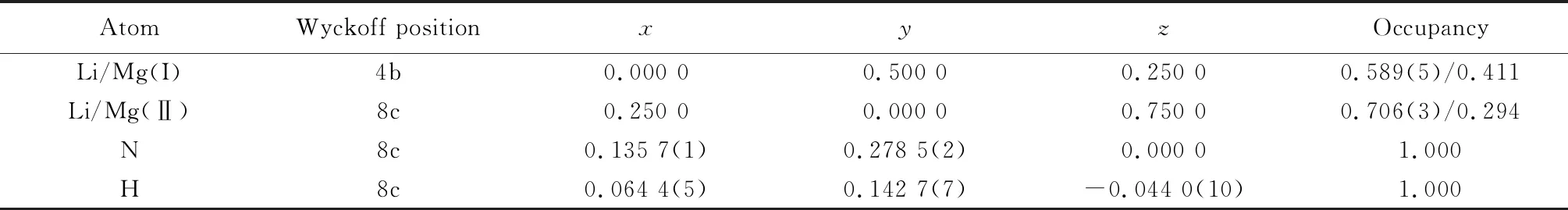
表1 α-Li2MgN2H2晶胞中各原子的具体占位信息Table 1 Specific occupancy information of each atom in the unit cell of α-Li2MgN2H2
Ti掺杂Li2MgN2H2时,因Ti和Li、Mg的原子半径相差不大,所以其倾向于替换Li或Mg的位置,形成Li7TiMg4N8H8或Li8Mg3TiN8H8[17]。通过分析α-Li2MgN2H2晶体结构中Li和Mg的占位可知,Ti替换Li或者Mg时存在图1中所示的8种情况。因此,本文将分别对Ti替换①~⑤位置的Li和⑥~⑧位置的Mg这8种晶体结构进行计算,详细分析Ti掺杂对Li2MgN2H2材料的生成焓、能带结构、态密度、差分电荷密度和布居等的影响情况。
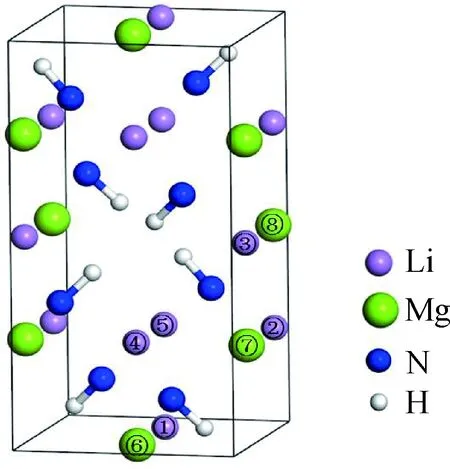
图1 α-Li2MgN2H2的晶体结构示意图Fig.1 Schematic diagram of the crystal structure of α-Li2MgN2H2
1.2 计算方法
计算采用Material Studio软件中的CASTEP程序[18]完成,采用基于密度泛函理论(DFT)[19]的第一性原理赝势平面波方法和周期性边界条件,晶体波函数由平面波基组展开,电子交换关联能函数采用广义梯度近似(GGA)中的PBE关系式[20],势函数采用倒空间表述的超软(ultrasoft)赝势[21]。平面波的截止能取为400 eV,简约布里渊区K点采用Monkhorst-Pack[22]方法选取,Ti掺杂前、后Li2MgN2H2的K点均选取为3×5×5。在进行计算前,采用BFGS方法[23]对各晶胞模型进行结构优化,确定它们的局域最稳定结构。自洽计算(SCF)时,应用Pulay密度混合法[24],并应用基集修正[25],自洽收敛条件为:总能量小于1.0×10-6eV/atom,每个原子上的应力低于0.003 eV/nm,公差偏移小于0.000 1 nm,应力偏差小于0.05 GPa。计算中各原子外层电子的电子组态分别为Li 1s22s1、Mg 2p63s2、N 2s22p3、H 1s1、Ti 3s23p63d24s2。
2 结果与讨论
2.1 杂质替换能
掺入杂质原子必然会影响Li2MgN2H2结构的稳定性,为了明确掺杂Ti在Li2MgN2H2结构中的稳定替代位置,首先计算了Ti替换不同位置的Li或Mg存在的8种不同情况下的Li2MgN2H2的杂质替换能,其定义公式如下[20]:
(1)
表2为Ti替换8个不同位置(如图1所示,分别标记为①~⑧)的Li或Mg后Li2MgN2H2体系的总能量和杂质替换能。可以看出,Ti掺杂Li2MgN2H2体系的杂质替换能均为负值,且Ti替换Mg的杂质替换能要比替换Li的杂质替换能小,说明掺杂反应均可进行,且Ti掺杂Li2MgN2H2体系时,其更倾向于替换Mg。进一步对比Ti替换不同位置的Mg后体系的杂质替换能发现,Ti替换位置⑦的Mg时,其替换能最低。因此,Ti掺杂Li2MgN2H2体系时,会优先替换位置⑦的Mg。

表2 Ti替换不同位置的Li或Mg后Li2MgN2H2体系的总能量和杂质替换能Table 2 Total energy and impurity replacement energy of the Li2MgN2H2 system after Ti replaces Li or Mg in different positions
本文将通过对比未掺杂的Li2MgN2H2体系和Ti替换位置⑦的Mg之后的Li8Mg3TiN8H8体系的结构参数、生成焓、能带结构、态密度、差分电荷密度及布居,详细研究Ti掺杂对Li2MgN2H2材料储氢性能的影响及作用机理。
2.2 结构优化
表3为未掺杂Li2MgN2H2和Ti掺杂Li2MgN2H2体系经结构优化后的晶胞参数。可以看出,Li2MgN2H2晶胞参数的计算值与实验值(a=0.978 71 nm,b=0.499 27 nm,c=0.520 19 nm)[12]吻合很好,说明本文所选计算参数及条件合理可靠,计算结果可信度高。此外,与未掺杂体系相比,Ti掺杂Li2MgN2H2体系的晶胞参数均有所增大,且其晶胞体积增大了0.007 61 nm3,说明Ti掺杂使得Li2MgN2H2晶胞中各原子间的间隙增大,有利于该材料在吸放氢过程中的氢气传递,从而使其吸放氢动力学性能得到改善。

表3 Ti掺杂前、后Li2MgN2H2的晶胞参数Table 3 Unit cell parameters of Li2MgN2H2 before and after Ti doping
2.3 生成焓
为了弄清Ti掺杂对Li2MgN2H2材料储氢性能的影响,接下来,从化学反应的角度对Ti掺杂前、后Li2MgN2H2体系的生成焓进行了计算,进而分析了掺杂前后体系的稳定性。
Li2MgN2H2晶胞中包含8个Li、4个Mg、8个N和8个H,其生成焓可采用式(2)进行计算:
ΔH={Etot(Li8Mg4N8H8)-8Etot(Li)-4Etot(Mg)-4Etot(N2)-4Etot(H2)}/4
(2)
式中:Etot(Li8Mg4N8H8)是Li2MgN2H2晶胞的总能量,为-7 726.82 eV;Etot(Li)、Etot(Mg)、Etot(N2)和Etot(H2)分别为单质Li、Mg、N2和H2的能量值,经计算,其具体数值分别为-189.97 eV、-973.95 eV、-541.31 eV和-31.70 eV。将上述各数值代入到式(2)中,计算得到Li2MgN2H2体系的生成焓为-463.93 kJ/mol。
Ti掺杂Li2MgN2H2的晶胞中含有8个Li、3个Mg、1个Ti、8个N和8个H,其生成焓可利用式(3)进行计算:
ΔH={Etot(Li8Mg3TiN8H8)-8Etot(Li)-3Etot(Mg)-Etot(Ti)-4Etot(N2)-4Etot(H2)}/4
(3)
式中:Etot(Li8Mg3TiN8H8)是Ti掺杂Li2MgN2H2晶胞的总能量,为-8 354.66 eV;Etot(Ti)是单质Ti的能量值,为-1 603.12 eV。代入到式(3)中,计算得到Ti掺杂Li2MgN2H2体系的生成焓为-431.75 kJ/mol。与未掺杂体系相比,Ti掺杂使得Li2MgN2H2体系的生成焓值降低了约32.18 kJ/mol,说明Ti掺杂能够有效地降低该体系的结构稳定性,有利于其吸氢反应的进行。
2.4 能带结构
为从电子结构的角度揭示Ti掺杂对Li2MgN2H2材料储氢性能的影响,在结构优化后,对Ti掺杂前、后的Li2MgN2H2沿布里渊区高对称方向的能带结构进行计算。其中,选取费米能级作为能量零点,且由于能带结构图中位于价带底部的能带并没有实际意义,因此着重对两者能带结构图中的价带和导带部分进行分析,结果分别如图2(a)和(b)所示。

图2 Ti掺杂前、后Li2MgN2H2的能带结构Fig.2 Energy band structure of Li2MgN2H2 before and after Ti doping
从图2(a)中可以看出,计算得到的Li2MgN2H2的能带结构与文献[16-17]研究结果较为一致。Li2MgN2H2的导带出现在2.22~8.16 eV的区域范围内,而价带出现在-2.76~0 eV之间,能隙约为2.22 eV。而且,其导带底和价带顶位于同一K点处,该材料表现出直接能隙半导体的特性。由图2(b)可知,Ti掺杂Li2MgN2H2的导带出现在-0.82~4.09 eV的区域范围内,而价带出现在-5.59~-2.82 eV之间,能隙约为2.00 eV。对比分析发现,Ti掺杂使得Li2MgN2H2的价带和导带均向低能量区域发生移动,且费米能级进入导带,材料表现出一定的金属性。此外,Ti掺杂使得Li2MgN2H2体系的能隙显著变窄,文献[26]指出能隙的大小可用来表征晶体结构稳定性的高低,能隙越大,则晶体结构稳定性越好,说明Ti掺杂可使得该体系稳定性降低,从而有利于其吸氢反应的进行。
2.5 态密度
进一步对Ti掺杂前、后Li2MgN2H2的总态密度和分波电子态密度进行计算,结果如图3(a)和(b)。从图3(a)中可以看出,Li2MgN2H2的总态密度在2.22~8.16 eV范围内的导带主要由Li 2s、Mg 3s、Mg 2p、N 2p和H 1s轨道的电子贡献,在-2.76~0 eV范围内的价带则主要是由Li 2s、Mg 3s、Mg 2p和N 2p轨道的电子贡献。值得注意的是,H 1s与N 2s、N 2p的分态密度均有大量重合,表明N和H以通过共价键形式结合而成的[NH]2-离子基团形成存在。由图3(b)可知,Ti掺杂Li2MgN2H2的总态密度在-0.82~4.09 eV范围内的导带主要由Li 2s、Mg 3s、Mg 2p、N 2p、H 1s、Ti 3p和Ti 3d轨道的电子贡献,在-5.59~-2.82 eV范围内的价带主要由Li 2s、Mg 3s、Mg 2p、N 2p、和Ti 3d轨道的电子贡献。
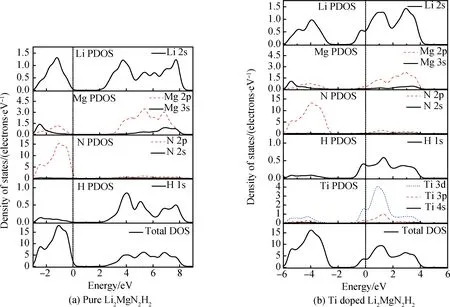
图3 Ti掺杂前、后Li2MgN2H2的总态密度和分波电子态密度Fig.3 Total and partial density of states of Li2MgN2H2 before and after Ti doping
与未掺杂体系对比,Ti掺杂后Li2MgN2H2的电子结构发生明显变化,且价带和导带均向低能量方向移动,这是由于过渡族金属Ti具有丰富的能级和特殊的3d电子结构,易产生多电子组态,在费米能级处的总态密度也主要是Ti 3d轨道的电子贡献。此外,Ti掺杂后体系中的Mg 2s和Mg 2p轨道的分波电子态密度的峰值均明显降低,这是由于Ti替换了Li2MgN2H2中的一个Mg,Mg原子贡献成键的原子个数由4个减为3个。进一步对比Li、N和H原子的分波态密度可知,Ti掺杂使得价带部分Li—N的成键峰和导带部分N—H键的成键峰峰值均有所降低,说明Ti掺杂可使体系中的Li—N和N—H键强减弱,从而有利于Li2MgN2H2材料吸氢反应的进行。
2.6 差分电荷密度及布居
为了更直观地理解Ti掺杂对Li2MgN2H2中各原子间成键情况的影响,进一步计算分析了Ti掺杂前、后Li2MgN2H2的差分电荷密度、键长和键布居。所选平面为取代原子位置与其最近邻的Li原子和N原子所构成的平面。

图4 Ti掺杂前、后Li2MgN2H2的差分电荷密度Fig.4 Differential charge density of Li2MgN2H2 before and after Ti doping
图4显示了Ti掺杂前、后Li2MgN2H2的差分电荷密度。从图4(a)中可以看出,在Li2MgN2H2中,N和H以共价键形式结合形成[NH]2-离子基团,而该基团与Li和Mg之间又分别以离子键相结合。从图4(b)中可以看出,用Ti替换Li2MgN2H2中的Mg1位置后,Ti1与N4之间的结合作用明显增强,从而使得N4—H4和Li5—N4有所减弱。这是因为,Ti作为过渡族金属元素,因含有空d轨道而表现出良好的电子亲和力(电负性:Ti>Mg>Li),可与N原子中的孤对电子发生作用,进而弱化N—H和Li—N键。
此外,通过电荷布居分析对体系中各原子间的结合键强弱进行了定量表征。表4列出了Ti掺杂前、后Li2MgN2H2的电荷布居数和键长。可以看出,Ti1替换Mg1之后,N4—Ti1之间的键布居数为0.91,键长为0.193 77 nm。与N4—Mg1相比,N4—Ti1的键长明显缩短,键强增大。此外,Ti掺杂使得N4—H4之间的键布居数由0.77降至0.53,键长由0.104 20 nm增长至0.109 66 nm,表明Ti掺杂使得N—H键强有所减弱。同时,Ti掺杂后Li5—N4之间的键布居数由0.04降至-0.24,表示原子之间形成反键,两原子之间相互排斥[27]。这与前面的差分电荷密度分析结果相吻合。
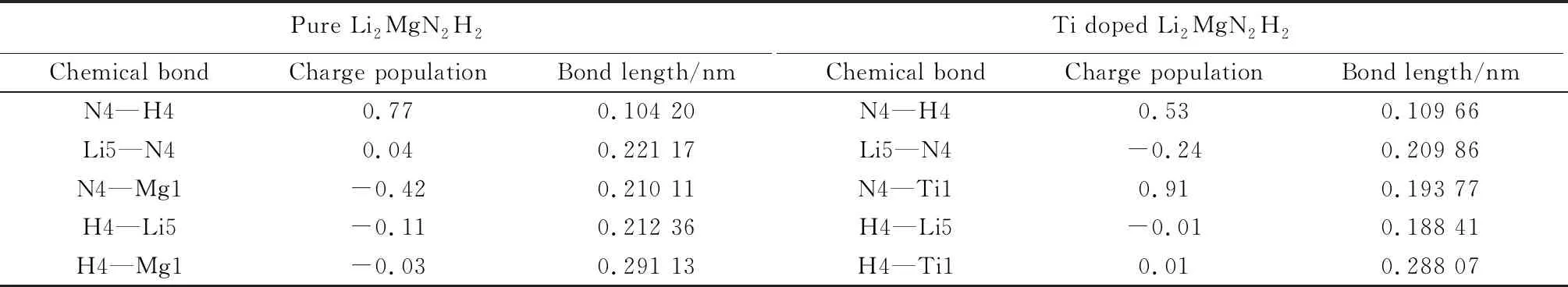
表4 Ti掺杂前、后Li2MgN2H2的电荷布居数和键长Table 4 Charge population and bond length of Li2MgN2H2 before and after Ti doping
同时,相关实验研究[11,17]指出,Ti基化合物掺杂可显著改善Li2MgN2H2材料的吸氢动力学性能,推测分析,这是因为Li2MgN2H2材料的吸氢动力学势垒主要来自N—H键和Li—N键断裂所需克服的能量,而过渡金属Ti外层的空轨道可有效吸引N原子外层的孤对电子,弱化了N—H键和Li—N键。这与本文计算分析结果相一致。
3 结 论
(1)Ti掺杂Li2MgN2H2体系时倾向于替换Mg,且当替换8c位置的Mg时,其杂质替换能最低,晶体结构最为稳定。
(2)Ti掺杂使Li2MgN2H2材料的生成焓值降低了约32.18 kJ/mol,降低了该材料的结构稳定性,有利于其吸氢反应的进行。
(3)Ti掺杂使得Li2MgN2H2的晶胞体积增大,能隙降低,且Ti与N原子之间较强的相互作用使得Li—N和N—H键的成键峰峰值降低,键强减弱。这些因素均有利于改善Li2MgN2H2材料的吸氢动力学性能。
