肝脏遗传性出血性毛细血管扩张症1例报告
2022-03-18王婷婷陈建平
王婷婷, 马 亮, 陈建平
江苏省常州市第一人民医院 消化内科, 江苏 常州 213003
1 病例资料
患者女性,50岁,因“体检行腹部超声发现肝脏血流异常”就诊本院。彩色多普勒血流显像:扩张的肝动脉及其分支内可见红蓝相间闪烁的明亮血流信号,且血流束较粗,大于正常同级的肝动脉内径(图1)。询问病史:患者无腹痛、腹胀,无眼黄、尿黄,无呕血、黑便,无纳差、乏力,无发热,无消瘦。体检:嘴唇、舌面、双手指掌面可见散在多处毛细血管扩张,直径2~4 mm,余未见异常。查血常规及肝肾功能均正常。进一步完善全腹增强CT+CTA示:肝右叶两枚囊肿,脾肿大;CTA示腹腔干及其分支增粗、扭曲,考虑遗传性出血性毛细血管扩张累及肝脏(图2、3)。追问病史:患者近数十年间无明显诱因反复鼻出血,量不多,可自行停止,一直未予诊治,患者两位同胞兄弟也有类似鼻出血病史。根据患者有长期反复鼻出血,嘴唇、舌面、双手指掌面可见散在多处毛细血管扩张及肝脏、腹部动脉多处受累,该患者诊断遗传性出血性毛细血管扩张症(hereditary hemorrhagic telangiectasia, HHT)。 鉴于患者临床仅表现为自发性少量鼻出血而无其他系统症状,无血红蛋白下降及肝功能异常,嘱其观察随访,定期复查腹部超声。
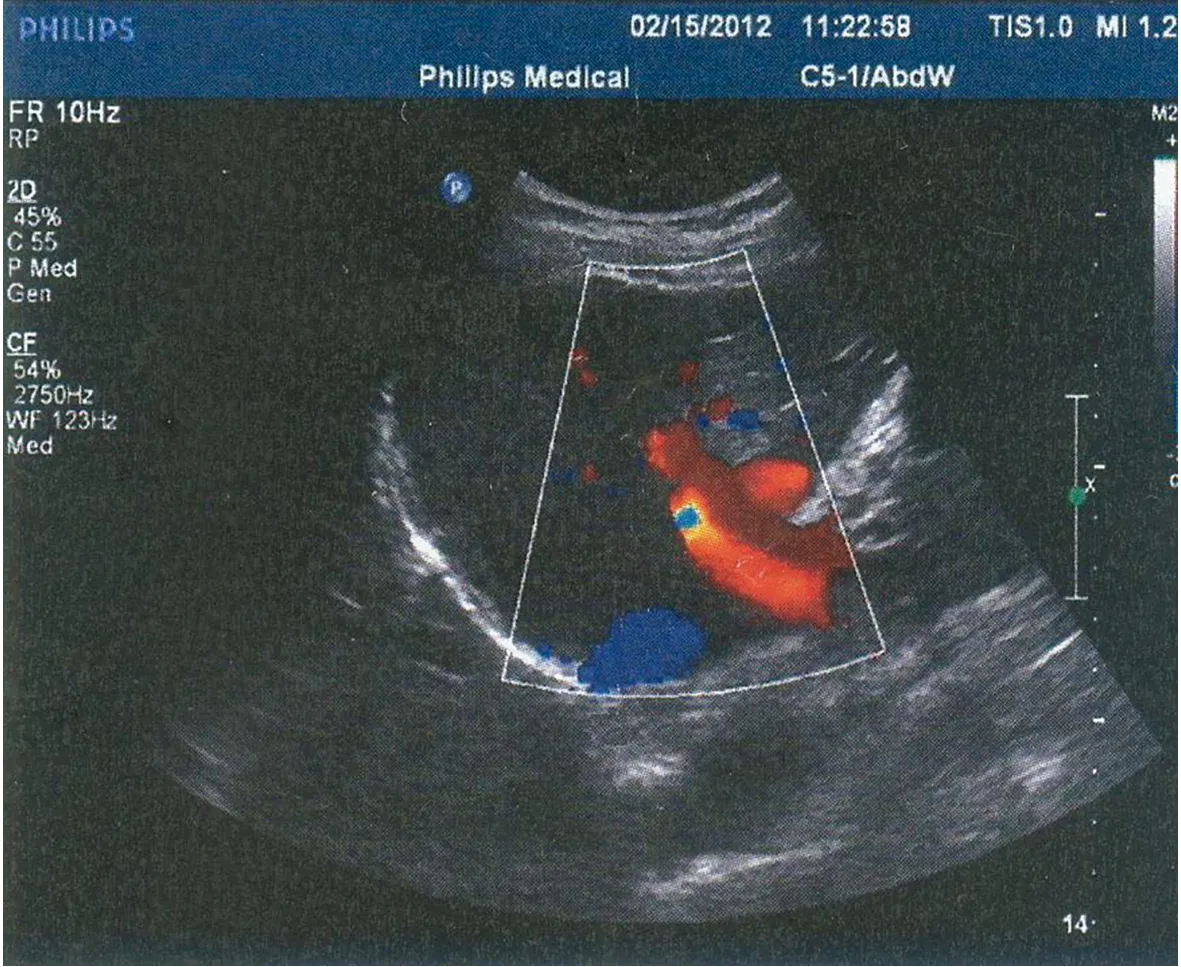
图1 彩色多普勒血流显像

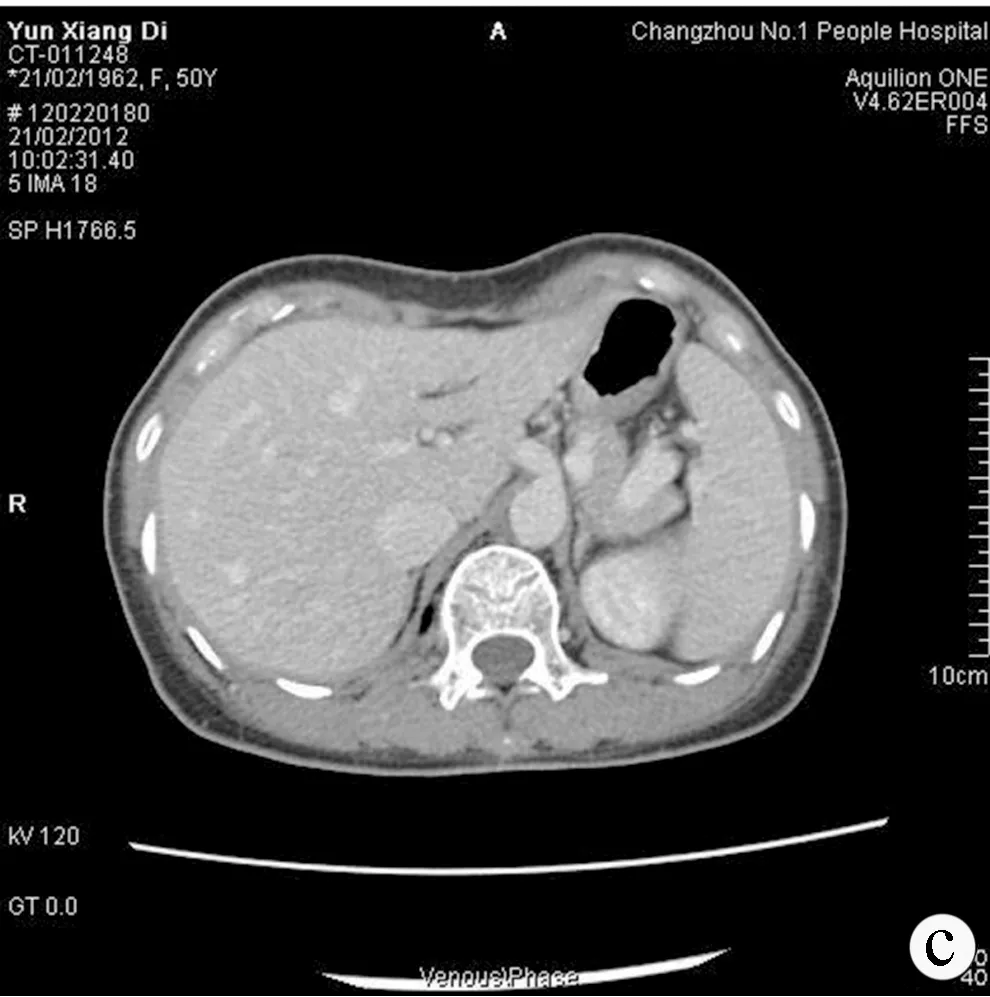
注:a,平扫期。肝脏、脾脏体积稍增大,肝脏右叶见数枚类圆形囊性低密度影,境界清、壁薄无强化;b,动脉期。肝内、肝门区可见粗细不均、走行迂曲的动脉血管,肝实质强化不均,门静脉及肝静脉部分显影;c,门静脉期。门静脉系统显影浅淡,脾静脉和下腔静脉增粗,主干迂曲扩张。

注:腹腔干及其分支增粗、扭曲。图3 腹主动脉CTA
2 讨论
HHT于1864年由Sutton首次报道,又被称为Rendu-Osler-Weber病,是由于血管系统发育异常导致毛细血管扩张和动静脉畸形的一种罕见的常染色体显性遗传病[1]。HHT世界范围内的发病率在1.5/10 000~1/5000,存在地域及种族差异,但无性别差异。欧洲发病率最高,特别是丹麦和法国,库拉索岛和博内尔岛的加勒比海人也有较高的发病率[2-3]。我国尚无HHT流行病学调查资料。HHT可累及鼻、口腔、舌、四肢等全身皮肤黏膜,以及肝、肺、脑、胃肠道等内脏器官,主要病理变化为毛细血管扩张和动静脉畸形(arteriovenous malformations,AVM)。反复自发性鼻出血是HHT最常见的临床表现。随着疾病进展,约15%的患者伴有胃肠道出血,并可能导致缺铁性贫血。动静脉畸形主要累及肺、脑和肝脏,发病率分别为15%~50%、25%和30%~80%,另外,脊髓动静脉畸形可见于儿童,容易导致截瘫[2]。
目前HHT的诊断仍采用库拉索标准:(1)鼻出血,呈自发性、反复性;(2)毛细血管扩张,位于多个特征性部位,如嘴唇、口腔、手指、鼻腔;(3)内脏损坏,胃肠道的毛细血管扩张,肺、肝、脑或脊髓的动静脉畸形;(4)家族史,一级亲属根据以上标准被诊断为HHT[4]。如果符合上述4项中3项及3项以上标准,则明确诊断为HHT;若符合2项标准,则为“可能或疑似”;如果少于2项,可以排除HHT。该患者符合前3项,因此诊断HHT明确。但采用库拉索标准时,临床医师需要考虑患者的年龄,因为HHT的症状和体征经常延迟出现。至少有90%的HHT患者直到40岁才符合HHT临床诊断标准,但在10岁时几乎没人符合,因此需要特别关注儿童和青少年漏诊的风险,对于这些群体,基因检测尤为适用[5]。
虽然HHT患者肝脏受累并不少见,但有临床症状的患者却不足10%,这些症状主要包括高输出量心衰竭(劳累性呼吸困难,端坐呼吸,水肿)、门静脉高压(静脉曲张破裂出血,腹水)、胆管系统(反复胆管炎,胆管坏死,肝脓肿)、门静脉系统脑病、盗血综合征(肠缺血)[2,5-6]。对怀疑肝脏受累的HHT患者,肝功能可出现异常,同时通过影像学检查可以明确诊断,这些检查方法包括超声多普勒、MRI、CT和肠系膜血管造影。其中超声检查能够用于肝受累HHT的诊断,是诊断HHT的首选筛查手段,肝动脉迂曲扩张半血流速度明显增快是肝受累HHT的特征性超声表现[1]。该患者是在体检时偶然发现,唯一线索为长期反复鼻出血,因而容易漏诊,但患者的彩色多普勒超声、腹部CT及CTA的描述符合上述肝脏HHT的影像学特征,有利于HHT的确诊。此外,由于HHT合并肺AVM、脑AVM可发生威胁生命的严重并发症如中风、短暂性脑缺血发作、脑脓肿、咯血、自发性血胸、颅内出血,因此早期筛查非常必要,推荐采用经胸腔的超声造影检查、颅脑MRI检查[5,7]。在本病例中,虽然患者无呼吸及中枢神经系统症状,但应建议患者行肺AVM和脑AVM的筛查,根据结果进一步咨询相关领域的专家。
对于无症状而影像学异常的肝脏HHT患者建议每3年复查1次超声;对于无症状而肝功能异常的患者则需每年复查1次[3]。对于有症状的肝脏HHT患者,根据不同情况可先选择内科强化对症治疗,若疗效不佳,特别是顽固的心衰、顽固的门静脉高压以及缺血性胆管坏死者需要考虑肝移植。肝移植是唯一具有确切疗效的治疗方案,其术后病死率为7%~10%,长期生存率为82%~92%[8]。另外,随着对HHT发病机制研究的深入,也为临床治疗提供了更多的可能性。1型HHT(HHT1)和2型HHT(HHT2)是HHT最常见的亚型,HHT1是由于编码内皮糖蛋白的基因发生突变[9],HHT2是由于编码活化素受体样激酶1的ACVRL1基因发生突变[10],两者占HHT患者的85%以上。另有近2%的患者是由于编码信号传导蛋白Smad4的SMAD4基因突变,它的突变可引起HHT伴幼年性息肉病综合征[7,11]。骨形态发生蛋白9基因突变所致的HHT相关疾病较为罕见。基因突变与生俱来,上述4种基因突变的检测可发现约85%的HHT患者具有致病基因突变。因此临床和基因检测相结合的综合诊断可以早期发现并诊断HHT[12]。上述基因的突变使血管壁弹力纤维及平滑肌缺乏,管壁变薄,完整性受损,导致毛细血管扩张、动静脉畸形[7,13]。血管生成的主要蛋白,血管内皮生长因子(vascular endothelial growth factor,VEGF)在HHT患者的血液和组织中高于正常人,进一步发现内皮糖蛋白、活化素受体样激酶1通过调控VEGF-VEGFR1/2参与到血管的生成及调节[7,14]。基于此,VEGF受体拮抗剂,贝伐单抗(Bevacizumab)运用于临床。贝伐单抗治疗鼻出血的临床研究数据较多,而关于其对肝脏HHT的治疗研究相对较少。临床有报道[15-16]静脉使用贝伐单抗(按照每2周5 mg/kg的剂量,共6个疗程)治疗HHT高输出量心衰竭及逆转肝移植的病例。另有一些抗血管生成药物如他克莫司、酪氨酸激酶抑制剂(索拉非尼、舒尼替尼、厄洛替尼、帕唑帕尼)以及基因治疗正处于细胞及动物实验研究阶段,未来或可运用于临床[7,17-18]。
HHT为进展性疾病,其外显性随年龄的增长而增加,早期积极、个体化治疗具有较好的临床效果。初始无症状或轻症肝脏受累的HHT患者可能迅速进展为肝硬化失代偿期、心衰竭或胆管坏死,故临床观察和随访具有重要意义,超声检查简便易行,是筛查的有效手段。因此临床及影像科医生应该提高对该病的认识,特别是对于无症状患者。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:王婷婷负责收集资料,撰写论文;马亮、陈建平负责指导修改文章并最后定稿。
