乙型肝炎病毒X基因及其变异促肝细胞癌的作用机制
2022-03-18周鑫宇柳东红蔡仕良陈宏森何奕达王瑞华曹广文
周鑫宇 柳东红 蔡仕良 陈宏森 何奕达 王瑞华 曹广文
据全球癌症统计数据,2020年原发性肝癌发病人数居所有癌症第六位,而死亡人数居所有癌症第三位;且在全球范围内,肝细胞癌(hepatocellular carcinoma,HCC)是原发性肝癌的主要类型,占 75%~85%[1-2]。在我国,原发性肝癌位于非成熟死亡(平均寿命之前的死亡)原因第一位,其中HCC占94.6%,且我国的HCC主要由乙型肝炎病毒(hepatitis B virus,HBV)慢性感染引起(占87.5%)[3-5]。尽管随着HBV疫苗的普及以及对携带者进行积极的抗病毒治疗,HBV感染所致的HCC造成的负担已逐渐减轻[6-8],但治疗乙型肝炎仍然是我国HCC初级预防的关键战略。
HBV致癌依赖三大因素:病毒复制、整合和进化,其中HBV X基因(HBV X gene,HBx)在这个过程中起主要作用。HBx通过促进共价闭合环状DNA(covalently closed circularDNA,cccDNA)结合的组蛋白乙酰化和病毒限制因子泛素化等机制促进HBV复制。HBx还是HBV整合到宿主基因组的最主要区域,并导致有促癌能力的C-端截短型HBx(c-terminal truncated HBx,Ct-HBx)和融合转录本形成。同时,HBx也是HBV进化的主要区域,免疫微环境和遗传选择下的HBx变异具有更强的致癌能力。此外,HBx及其变异可以调控关键基因的表达,影响相关信号通路激活,从而发挥原癌基因作用,进而参与HCC发生发展中的多种生物学过程[9]。因此,研究HBx及其变异在HBV相关HCC(HBV-HCC)发生发展以及防治等方面的作用有重要意义。本文就HBx及其变异在HCC中的致癌作用及其机制进行综述。
1 HBx调控HBV复制
HBV是一种部分双链的环状DNA病毒,约3.2 kb。HBV病毒具有4个开放阅读框,分别是S区、C区、P区以及X区,编码7种蛋白质(preS1、preS2、S、preC、C、病毒聚合酶、HBx蛋白)和4个调控元件(增强子Ⅱ/基础核心启动子、preS1启动子、preS2/S启动子和增强子Ⅰ/X启动子)。HBx基因全长为779 bp(nt.1060~1838),其中nt.1060~1373段为HBx的启动子区,nt.1374~1838为HBx的编码区;而HBx的编码区是HBV基因组最小的开放阅读框,编码154个氨基酸,蛋白大小为17 kDa;因HBx蛋白与目前已知的蛋白均不具有同源性,故而得名[9]。HBx促癌机制见图1。
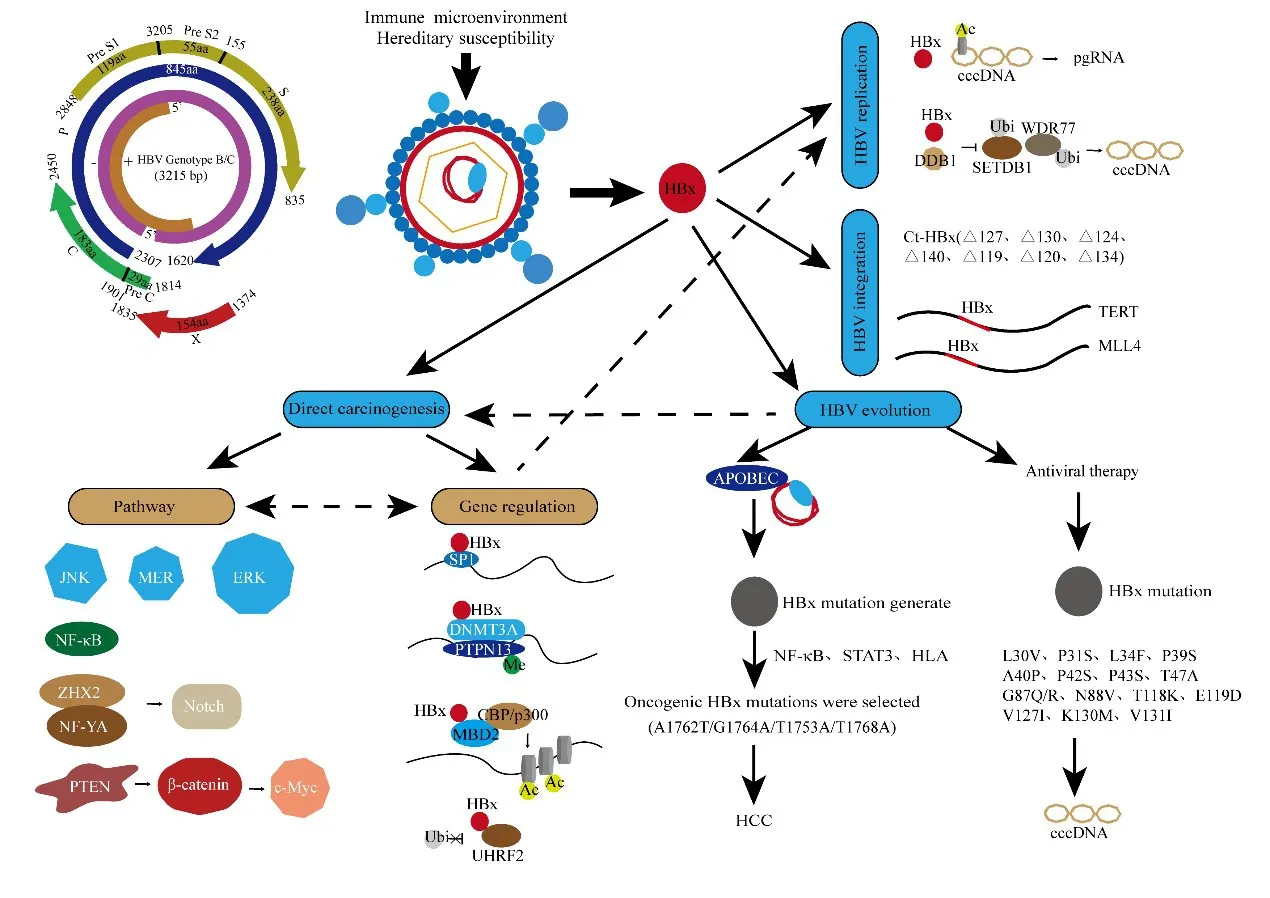
图1 HBx促癌机制示意图Fig.1 Schematic diagram of HBx promoting cancer mechanism
HBV的复制可导致持续的慢性炎症,这也是导致肝硬化和HCC发生的关键因素,高病毒载量还与不良预后相关[10]。在HBV的生命周期中,HBx是HBV cccDNA转录和病毒复制的调控蛋白。HBx通过参与多种表观遗传修饰如甲基化、乙酰化、泛素化和非编码RNA的表达来影响cccDNA的表达和HBV复制[11]。
乙酰转移酶和去乙酰转移酶通过调控cccDNA结合的组蛋白状态,对cccDNA发挥翻译后修饰功能,从而影响HBV复制。HBx在促进cccDNA结合的组蛋白乙酰化的同时也防止其去乙酰化,从而产生更多的前基因组RNA,并增加HBV循环[12]。HBx还可使关键分子泛素化,干扰天然免疫反应对HBV的攻击,从而实现免疫逃逸并维持HBV稳定复制。DNA损伤特异性结合蛋白1(damage-specific DNA binding protein 1,DDB1)是病毒复制必须的,HBx与DDB1的相互作用还可使HBV限制因子WDR77、SMC5/6和SETDB1等泛素化和降解,从而促进cccDNA转录和HBV复制[13]。miRNA可以调控HBV复制,同时也能被HBV复制所调控,其中HBx起主要调控作用。HBx引起的miR-122下调还会增强HBV的复制能力,同时诱导病毒持续性感染[11]。
2 HBx是HBV整合的高频区域
HBV整合到宿主基因组是HCC发生的分子机制之一,高通量测序表明,在76.9%的HBV-HCC患者中,HBV基因组会整合到宿主基因中[14]。虽然HBV是随机整合到宿主基因组中,但是病毒基因组内nt.1600~nt.1900之间的区域(对应HBx区的3'端和前核心区的5'端)是发生整合最常见的区域[15]。
2.1 Ct-HBx具有更强的促癌能力
HBx区的3'端整合到宿主基因组导致Ct-HBx产生,且Ct-HBx与不良预后相关[16]。目前,各种不同的Ct-HBx已在HBV感染以及相应的肝硬化和HCC患者中被发现。Ct-HBx较全长HBx有更强的促癌作用,其中与HCC发生发展关系密切的Ct-HBx包括HBx-△127、HBx-△130、HBx-△124、HBx-△140、HBx-△119、HBx-△120、HBx-△134等[17]。其中研究最多的是HBx-△127,其可促进正常肝细胞中NF-κB、生存素和人类端粒酶逆转录酶活性,以及促进c-Myc和增殖细胞核抗原表达水平,还能通过5-LOX上调SREBP1c表达增加FAS的转录,从而促进肝癌细胞生长[18]。HBx-△120或HBx-△134可与NFATC2协同作用下调TXNIP表达,从而导致糖代谢重编程,进而启动HCC的发生并促进癌细胞迁移和侵袭[19]。HBx-△130或HBx-△119可以激活 caveolin-1/LRP6/β-catenin/FRMD5轴,从而促进HCC发生及侵袭、转移[20]。
2.2 HBx导致宿主基因组整合突变
HBV整合到宿主基因组上,从而干扰宿主基因组稳定性。HBx整合到TERT、MLL4和FN1等癌症相关基因的调控区域并影响其表达[15]。HBV整合容易产生HBV-宿主嵌合转录本,发挥致癌作用。HBV-宿主嵌合转录本主要是HBx基因与人类基因内含子内重复元件的融合[21]。HBx-LINE1嵌合转录本作为一种长链非编码RNA,可下调miR-122表达水平,导致WNT/β-catenin通路活性增加,并诱导集落形成以及细胞侵袭和迁移,最后导致肿瘤发生发展[22]。
3 HBV进化产生更强致癌能力的HBx变异
在慢性乙型肝炎进展到HCC的过程中,HBV进化是最突出的分子事件[23-24]。HBV的进化遵循达尔文模型:突变-选择-适应。在HBV病毒复制过程中因缺乏校对功能而容易发生基因突变,而慢性炎症环境使突变率增加[23,25]。大多数HBV突变体被抗病毒免疫反应消除,只有小部分利于病毒复制生存且促进肝细胞再生的突变HBV能够存活,并逐渐发展成促HCC克隆[26]。HBx是HBV发生突变的高频区域,在人载脂蛋白B mRNA编辑酶催化多肽(apolipoprotein B mRNA-editing enzyme catalytic polypeptides,APOBECs)、遗传易感性、免疫微环境和抗病毒治疗等作用下,HBx变异发生累积,而具有强烈致癌作用的突变型HBx被选择,从而可以预测HCC的发生与复发。
3.1 HBx变异的产生与APOBECs
使HCC发生风险增加的HBV突变主要位于增强子Ⅱ/基础核心启动子区/前核心(EnhⅡ/BCP/PC)以及preS区域[27]。其中增强子Ⅱ(nt.1636~1744)和基础核心启动子区(nt.1751~1769)与HBx开放阅读框(nt.1060~1838)重叠。其中,APOBECs诱导的突变是HBV变异的来源[28]。APOBEC3s的表达水平与HBV准种复杂性呈正相关[29]。衰老、环境暴露和遗传易感性有助于激活和维持非溶解性炎症,活化的炎症信号通路可以反式激活APOBECs表达,从而促进HBV和体细胞突变。IL-6诱导的APOBEC3B-UNG失衡的遗传多态性能显著促进HCC-HBV突变的产生[30]。EnhⅡ/BCP/PC区对APOBEC3成员的编辑非常敏感,编码HBx的序列尤其容易被编辑。APOBEC3更倾向于将HBx区域作为其编辑靶点,并产生Ct-HBx等突变型[31]。
3.2 HBx突变在HCC发生发展过程中逐渐累积
目前在病例对照研究和队列研究中均发现位于HBx基因区的几个关键突变(G1613A、C1653T、T1674C/G、T1753V、A1762T/G1764A)是HCC发生的风 险 指 标[32-33],突 变 组 合(C1653T、T1753V 和A1762T/G1764A)还可有效预测HCC的发生[34]。在一项包含43项研究的荟萃分析中发现,C1653T、T1753V以及A1762T/G1764A双突变均与HCC发生风险有关,且这些突变在乙型肝炎感染者从无症状携带发展为HCC过程中是逐渐累积的,同时无论是存在单独位点还是这些位点的组合突变都具有HCC发生风险[35]。以上流行病学研究表明,在慢性肝炎所致的肝硬化向HCC进展过程中,HBx突变的种类和频率逐步累积,且HBx突变的累积是HCC发生的高危因素,HBx突变组合可用于预测HCC的发生和发展。
3.3 HBx突变与遗传及免疫环境相互作用
HBx突变的选择是由遗传及免疫微环境共同决定的,与欧洲人群相比,我国人群的常见基因型与HBV慢性感染风险增加以及HBx突变的免疫选择显著相关[26]。在我国人群中,STAT3基因的rs2293152少见基因型显著促进HBx突变A1762T/G1764A和T1674C/G的选择,并且与HBx突变存在显著的交互作用,协同促进HCC发生[36]。NF-κB1与其抑制分子NFKBIA启动子遗传多态性是HBV C型感染者发生HCC的危险因素,NFKBIA启动子SNP,rs2233406和rs3138053的少见基因型(rs2233406T、rs3138053G)通过降低NFKBIA表达,增加NF-κB1活性,促进炎症对A1762T/G1764A、T1753V等促癌HBx突变的选择[37]。rs28362491的变异基因型(缺失型)与HBV C型感染者中A1762T/G1764A频率增加显著相关;rs2233406少见基因型还与A1762T/G1764A交互作用而显著增加HCC发生风险[37]。HLA-DP的遗传多态性(rs3077、rs3135021、rs9277535)与HBV炎症慢性化密切相关,促进 HBV 清 除 的 基 因 型(rs3077T、rs3135021A、rs9277535A)与HBx促癌突变(C1653T、T1674C/G等)呈负相关,而与抑癌HBx突变(G1652A、T1673C、T1674C、G1719T、G1730C、G1799C 和A1727T)呈正相关[38]。我国HLA-DR SNPs少见基因型(rs3135395T、rs3135338C和rs477515T)可显著降低HBV持续存在和HCC发生风险,且HLA-DR少见基因型与HBx区突变(A1762T/G1764A、T1753V和C1653T)的产生呈负相关[39]。还有研究显示,HLA-DP、HLA-DR和HLA-DQ的遗传多态性均与HBx突变在乙型肝炎导致的HCC中存在显著的交互作用[40]。
3.4 HBx突变与抗病毒治疗药物
高病毒载量与HBV-HCC患者术后不良预后相关,抗病毒治疗可以显著降低HCC复发及其相关死亡风险[41-42],但对Ct-HBx的促癌能力没有影响[43]。长期治疗期间,核苷类似物的疗效会因耐药突变体的出现而降低。研究发现在耐药患者中HBx的L30V、P31S、L34F、P39S、A40P、P42S、P43S、T47A、G87Q/R、N88V、T118K、E119D、V127I、K130M、V131I等15个氨基酸残基出现了较高的突变频率,还存在多个位点的缺失/截断和插入,且这些耐药患者中的HBx突变通过增加核cccDNA水平补偿核苷类似物对HBV复制的抑制作用[44]。
3.5 HBx突变影响HCC发生发展的机制
HBx基因区的碱基突变可引起对应的氨基酸改变,影响HBx蛋白的结构和功能,从而增强HBx的致癌潜能和HBV复制和免疫逃逸能力。A1762T和G1764A突变导致的HBx蛋白K130M和V131I突变是最常见的HBx突变类型,这两个突变常常一起出现,该类型突变可影响细胞周期调控基因的表达,促进细胞增殖和HCC的发生[45]。K130M/V131I突变,尤其是A1762T/G1764A/T1753A/T1768A组合突变(I127N/K130M/V131I/F132Y)可通过激活E2F1转录因子增强SKP2的转录,促进细胞周期抑制相关蛋白(p21、p53等)泛素化,从而促进细胞增殖[46]。在动物模型中K130M/V131I突变可通过激活AKT/FOXO1通路和影响花生四烯酸代谢诱导强烈的炎症反应,从而促进HCC进展[47]。野生型HBV可以激活HIF-1α而促进HCC的发生和发展,而K130M/V131I突变可以进一步上调HIF-1α的表达和转录活性[48]。K130M、V131I、K130M/V131I尤其是I127N/K130M/V131I/F132Y对细胞的迁移、增殖能力和Wnt/β-catenin信号通路的激活作用较野生型强[49]。
4 HBx影响信号通路和基因表达而直接致癌
HBx除了在HBV的复制、整合和进化中起关键作用,同时也作为一种重要的癌蛋白,参与多种HCC相关信号通路,影响关键基因的表达调控,从而促进细胞增殖、迁移、侵袭、耐药及诱导上皮间充质转化(EMT)和癌细胞干性,扰乱细胞代谢、影响DNA损伤修复等,发挥直接致癌作用[9]。
4.1 HBx参与的HCC信号通路
HBx在HCC发生发展过程中参与多条信号通路,其中备受关注的信号通路包括MAPK信号通路、NF-κB信号通路、Notch信号通路以及Wnt/β-catenin信号通路。HBx激活JNK信号通路,使PXN 178位点的丝氨酸磷酸化,从而促进HCC侵袭并导致不良预后[50]。lncRNA IHS同时受HBx和SMYD3调控,通过MEK/ERK信号通路促进HCC细胞迁移、侵袭和增殖[51]。HBx也能通过抑制miR-34a促进IKKβ诱导的NF-κB活化,以及诱导STAT3磷酸化,共同促进HMGB1表达和分泌,从而促进HCC的EMT进展和血管生成[52]。HBx还可通过激活HCC中的NF-κB信号通路上调CEBP/α,进而促进IL-34表达,最终导致HCC细胞增殖和迁移[53]。ZHX2与NF-YA相互作用能抑制Notch1转录,而HBx与转录因子CREB相互作用可促进miR-3188转录,下调ZHX2,降低ZHX2对Notch信号通路的抑制作用,从而诱导HCC的发生[54]。HBx还能下调PTEN而增强β-catenin/c-Myc信号通路,激活B7-H1的转录活性,增加PD-L1表达,抑制T细胞应答,促进HBV免疫逃逸[55]。HBx诱导的miR-5188也能损害FOXO1,从而刺激β-catenin核易位,激活Wnt信号通路,进而诱导肿瘤干细胞形成[56]。
4.2 HBx对基因表达的调控作用
4.2.1 HBx与转录因子相互作用 HBx蛋白是一种反式激活因子,但HBx本身不能直接与DNA结合,其转录活性是通过与多种转录因子相互作用,激活各种病毒和细胞的启动子和增强子,进而影响HCC的发生发展。如HBx与基础转录因子SP1协同作用,上调SWELL1[57]。HBx还可通过转录因子c-Jun激活LASP-1启动子活性,促进其表达[58]。
4.2.2 HBx与表观遗传修饰 表观遗传变化可影响编码和非编码基因的表达,促进HBV复制和HCC的发生发展。表观遗传改变可由多种因素引起,如病原体、化学物质、紫外线等。在HBV-HCC中,HBx被认为是影响表观遗传最重要的因素之一。HBx通过调节基因启动子的甲基化状态来调节下游分子表达。一方面,HBx上调DNMT3A表达并与DNMT3A相互作用,使PTPN13启动子区DNA甲基化水平升高,从而抑制PTPN13的转录[59]。另一方面,HBx可诱导RelA与EZH2、TET2和DNMT3L形成复合物,导致EpCAM的NF-κb侧CpG位点去甲基化,从而上调EpCAM表达[60]。HBx与MBD2以及CBP/p300相互作用可诱导MBD2-HBx-CBP/p300复合物形成,介导组蛋白H3和H4的乙酰化[61]。ZHX2是HCC相关肿瘤抑制基因,而HBx可促进 miR-155表达[62],进而降低ZHX2水平。DLEU2是一种lncRNA,HBx可增强其转录并诱导其在感染肝细胞中积累,HBx与DLEU2以及EZH2结合,可解除EZH2对其靶基因的抑制作用,促进病毒复制以及下游宿主基因的转录激活[63]。HBx与SKP2结合也可导致SHIP2泛素化,促进HCC进展[64]。
5 小结与展望
HBx基因与其编码的蛋白通过促进HBV复制、整合到宿主基因组和发生变异,并调控关键基因表达和信号通路的激活,广泛参与到慢性肝炎-肝硬化-肝癌的发生发展过程中,在HBV-HCC发生、复发、侵袭转移、免疫逃逸中起重要作用。目前,HBx对HBV复制、基因表达调控等方面已经得到广泛的研究,流行病学证据表明突变型HBx与HBV-HCC的关系密切。突变型HBx具有比野生型HBx更强的致癌能力且与抗病毒治疗耐药相关,但突变型HBx在HBV-HCC发生发展以及耐药过程中的具体作用机制研究仍较少。今后进一步深入研究HBx及其突变的致癌机制及研发靶向HBx突变的药物,对HBV-HCC的预防、治疗和预后预测有重要意义。
