High Energy Density Materials based on N4 Molecules:Synthesis Routes from First-principles Calculations
2022-03-18MAOYutingSUNChuliDUHuifangGUOWei
MAO Yu-ting,SUN Chu-li,DU Hui-fang,GUO Wei,2
(1.Key Laboratory of Advanced Optoelectronic Quantum Architecture and Measurement (MOE),School of Physics,Beijing Institute of Technology,Beijing 100081,China;2.Frontiers Science Center for High Energy Materials (MOE),Beijing Institute of Technology,Beijing 100081,China)
Abstract:To explore the routes for synthesizing and stabilizing polymeric nitrogen in molecules or crystals,two methods for synthesizing high energy density materials (HEDM)based on N4 molecules were proposed based on the first-principles calculations.The first route is to adsorb N4(Td)molecule on single crystal metal surfaces,and the second method is to use structural search to obtain stable N4-related structures.Three N4-containing crystals,P-43m-N4,P4/m-LiN4 and Amm2-G/N4 were proposed,and their energy densities,thermodynamic and kinetic stabilities,as well as their electronic properties were calculated.The results show that under ultra-high vacuum and low temperature conditions,N4(Td)can be stabilized by losing the Td symmetry on the metal surfaces.AIMD simulations indicate that they are stable at 50GPa and 50 or 300K.Through coordination bond between N4 and metal atoms,polymerized N4 can be synthesized at high pressure and stabilized at low temperature conditions.
Keywords:first-principles calculations;high energy density materials;N4;coordination bond
Introduction
In last two decades,various theoretical and experimental works have been done to investigate the synthesis route of N4.Lee et al.reported that the most likely synthesis route for N4(Td)comes from the association of two bound quintet states of excited N2molecules[12].Bittererová et al.proposed that N4(Td)can be formed from azide (N3)and an atomic N(N+N3→N4)[13].Legare et al.reported that two or more dinitrogen (N2)naturally occur coupling under ionospheric radiation conditions,forming a complex of[N4]2-chains connecting two boron centers[14-15].The methods of ionization and electrical discharge can only produce short-lived metastable molecules or ions[16-18].Meanwhile,researchers started with hybrid materials which are consisted of polymeric nitrogen and nanomaterials (such as carbon nanotubes,fullerenes,and graphene substrates)[19-27].In these hybrid materials,polymeric nitrogen chain or compound can be stabilized due to interaction with the surrounding or under high pressures.Such nitrogen-based HEDM needs to activate N2and stabilize the final product.However,it is rather challenging to synthesize stabilized hybrid polymeric nitrogen HEDM on a large scale because of the large energy difference between N2and N4.On the other hand,there have been great progress in the study of metal nitride clusters,where Li,V and Ti atoms coordinate with N atoms and form charged clusters by laser ablation technique[31-33].
In this article,based on first-principles calculation and ab initio molecular dynamic simulation (AIMD),we propose two routes to reduce the energy difference between N4and N2molecules.The first route is to use a transition metal surface to adsorb and stabilize N4at low temperature.We show that if produced hot N4(Td)molecule is dosed into ultra-high vacuum and adsorbed onto transition metal surfaces (such as a cluster of six Co adatoms on Pt(111)surface denoted as 6Co-Pt(111)in this work)at low temperatures,it can be stable after relaxation and losing its Tdsymmetry.The second route is to stabilize N4at high pressure in crystal,and three crystals containing N4are proposed in this study,which are P4/m-LiN4,P-43m-N4and G/N4.High pressure reduces the enthalpy difference between N4and 2N2,making the crystal thermodynamically stable.AIMD simulations indicate that such high nitrogen content HEDM are stable at 50GPa and 50 or 300K.
1 Calculation Models and Method
To stabilize the N4molecule,we have proposed two classes of configurations,which are N4/metal-surface adsorption configuration (metal=Ni,Cu,6Co-Pt)[31]and three molecular crystal structures.We first performed first-principles calculation based structural searching under high pressure with CALYPSO,which is a structural evolution code based on particle swarm optimization technique[35-36].We set the pressures to 20—100GPa to promote the polymerization of nitrogen atoms,then we constructed N4structure in the crystal and minimized the crystal with respect to unit-cell size and shape,as well as the atomic coordinates in the unit-cell.The first proposed crystal structure is P-43m-N4in cubic lattice with a N4(Td)molecule in each unit-cell,which is obtained by relaxing a CALYPSO predicted metastable structure in terms of lattice parameter and atom positions under pressure.The second structure is P4/m-LiN4in tetragonal lattice,which is comprised of one Li atom and one N4(D4h)ring in the unit-cell.Alkali metal azide has attracted more attention in recent years due to its unique physical and chemical properties[37-40].Based on the structure of NaN3and LiN3,we replaced N3with N4and optimized the structure to find the P4/m-LiN4structure.The third structure is Amm2-G/N4,which is made up of graphene layers and N4(Td)molecule in orthorhombic lattice.We found a CALYPSO predicted metastable structure with N3ring in between bilayer graphene,then we replaced the N3with N4molecule to build the Amm2-G/N4crystal.All the lattice information is listed in Table 1.

Table 1 Structure (at zero pressure)and calculation details
DFT calculations were performed by using the Vienna ab initio simulation package (VASP)program[37].The surface of the Brillouin area was covered by Monhorst-Pack grid.RPBE (Revised Perdew-Burke-Ernzerhof)[42]was used to describe the exchange-correlation effects when calculating N4adsorption on transition metal surfaces,since RPBE gives more accurate binding energy in molecule/transition metal system,and PBE[43](Perdew-Burke-Ernzerhof)was used for other systems.Pseudopotentials generated with PAW (projector-augmented wave)method were used in treating the valence electron-ion interaction[44].The convergence criteria of self-consistent electronic and ionic loops were set to 10-4eV and 0.05eV/Å,respectively.We set the cut-off energy of plane-wave basis to 450eV for N4/metal-surface (Ni,Cu,6Co-Pt),and Amm2-G/N4,550eV for P4/m-LiN4and P-43m-N4.The mesh sampling size,unit-cell volume and number of atoms of those structures are listed in Table 1 of the Supplementary Materials.Transition states of N4dissociation are calculated by CI-NEB method[45].Phonon frequencies were calculated using the supercell method implemented in the PHONOPY code[45],3×3×3 supercells were built for P-43m-N4,2×2×2 for P4/m-LiN4and Amm2-G/N4.AIMD in NpT ensemble were performed in the VASP code using a Langevin thermostat in 3×3×3 supercells for P-43m-N4,2×2×2 for Amm2-G/N4and 2×2×4 for P4/m-LiN4at a temperature of 50K or 300K,the time step was set to 1fs,and the pressures were set to 0GPa or 50GPa.We obtained the phonon vibration frequency through the finite displacement method,then calculated the polarizability of eigenvector of the Γ-point phonons to get off-resonance Raman activity of each mode using vasp_raman.py script[47].
2 Results and Discussions
2.1 N4 molecule on surfaces
For gas phase N4molecules,we have focused on the structures with the highest symmetry and lowest energy[4],which are N4(Td)and N4(D2h)as shown in Fig.1.
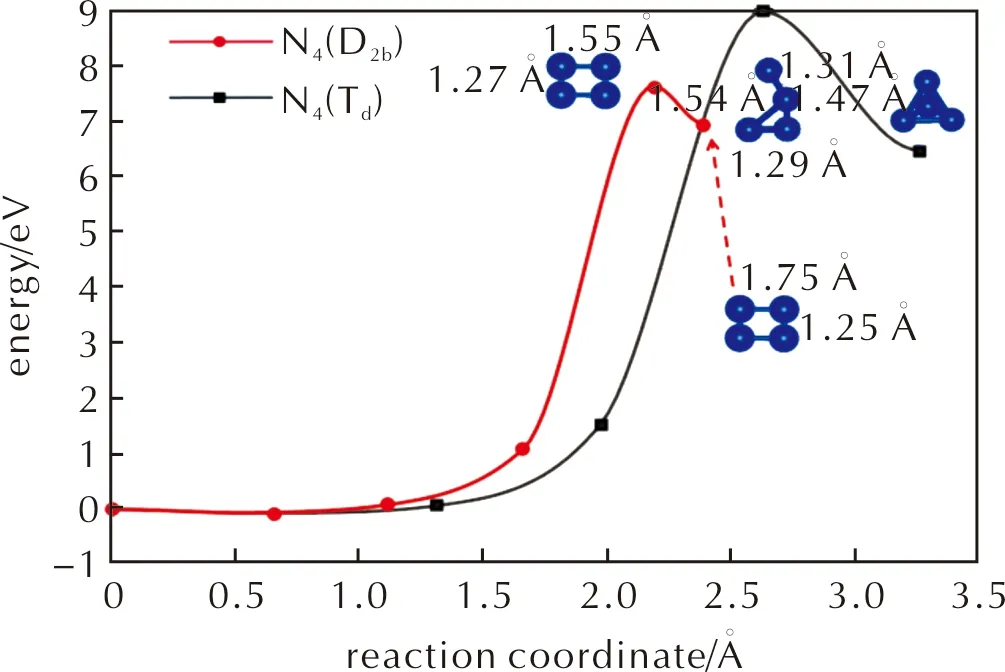
Fig.1 Potential energy diagram of 2N2→N4 reaction in gas phase. The blue balls are N atoms. The energy reference is set to two N2 molecules in gas phase.
It is rather difficult to synthesize N4molecule directly from N2molecules in gas phase,according to the calculated energy diagram in Fig.1.Synthesizing N4molecule is strongly endothermic (above 6.94eV for N4(D2h)and 6.48eV for N4(Td)),and the activation barriers are 7.61 and 8.97eV,respectively.Even if two N2molecules are activated and surpass the barrier to form N4,the decomposition barrier of N4molecule back to two N2is only 0.67 and 2.49eV for N4(D2h)and N4(Td)phases,respectively.Once they are prepared,via techniques such as microwave or electric discharge[13-18].N4(Td)should be more stable than N4(D2h)in gas phase.However,its lifetime is short,which is in a level of microsecond[13].
It is essential to stabilize N4molecule once it is produced and quenched at low temperature.Here we have proposed a few approaches to dissipate the excessive energy and stabilize N4molecules,so that further experimental techniques can be used to study its property.Transition metal surfaces often show good catalytic effect in many reactions due to proper binding strength to the intermediates in certain reactions[48].It is well known that N2is physisorbed on Pt (111),the binding is stronger at step sites,such as on Pt(211)surface[47],[50],and it can be even stronger when N2resides at step edges of nanoclusters on Pt(111),for example on 6Ni-Pt(111)surface (6 Ni adatoms form a cluster on Pt(111)surface),the adsorption can be enhanced and well understood by the d-band model[48],[51].


Fig.2 (a)N4 on Ni(111)surface. (b)N4 on Cu(111)surface. (c)Four different adsorption configurations (B,C,D,E)of N4 on 6Co-Pt(111)surface. Here,A represents two N2 molecular adsorbed on the 6Co cluster of the surface,which is taken as the reference state in panel (c). Red columns are the ΔE of configurations B,C,D,and E with respect to A.

Table 2 Bond length information
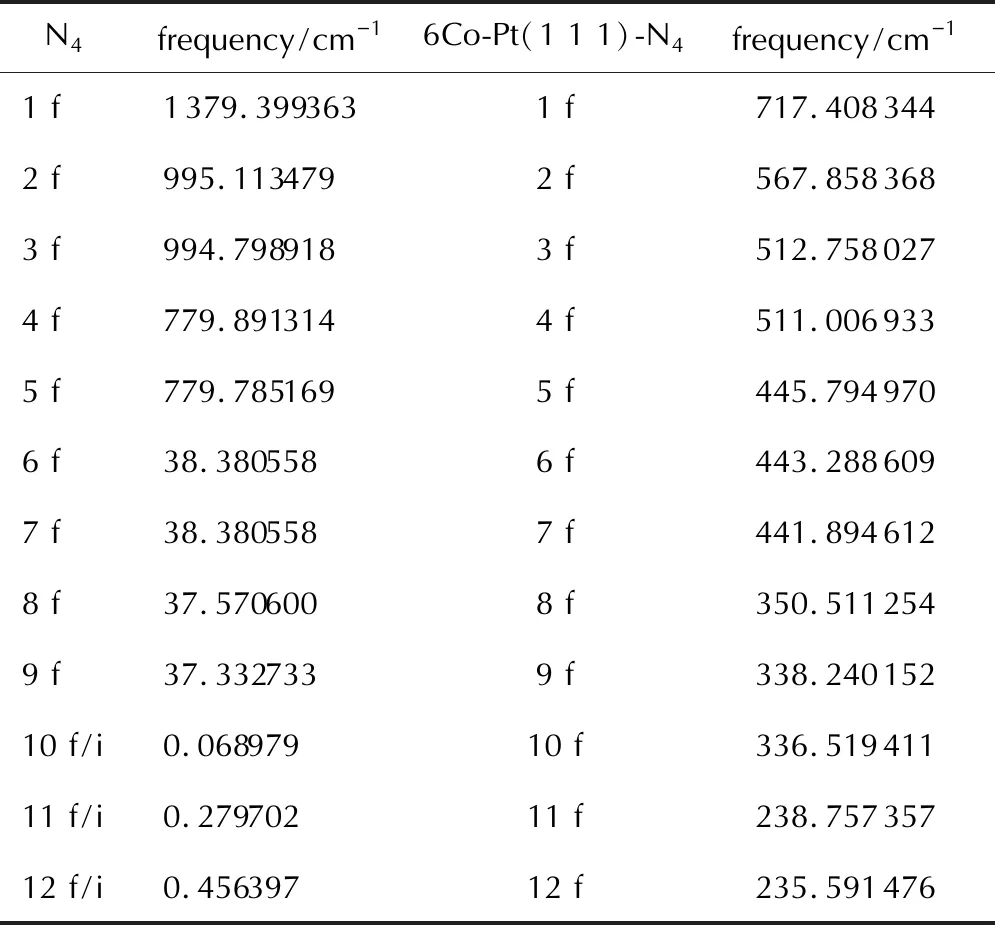
Table 3 The frequencies of N4 in the gas phase and N4 on the metal surface
2.2 N4 molecule in crystals
Another approach to stabilize N4molecule is to polymerize N atoms under high pressure.Here,we have proposed three crystal structures associated with N4,and the enthalpy differences as a function of pressure of the crystal with respect to their corresponding decomposition products are shown in Fig.3.
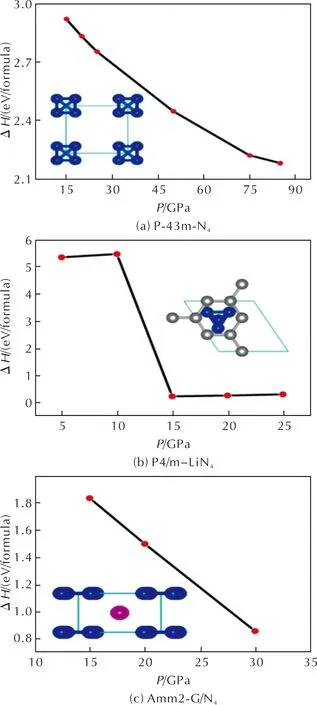
Fig.3 Enthalpy changes as a function of pressure. (a)ΔH=H(P-43m-N4)-1/2H(ɑ-N2). (b)ΔH=H(P4/m-LiN4)-1/2H(ɑ-N2)-H(LiN3). (c)The reference state is two N2 molecules in graphene layers. ΔH=H(Amm2-G/N4)-H(G/2N2).
The enthalpy difference decreases as the pressure increases,indicating that the crystal becomes thermodynamically more stable at higher pressures.Such trend implies that it is possible to synthesize N4containing crystals from N2under high pressure and low temperature conditions.Lattice parameters of the three crystals at different pressures are shown in Table 4,5 and 6 in Supplementary Materials.Next,we focus on the properties of such high-pressure phases of N4containing crystals.
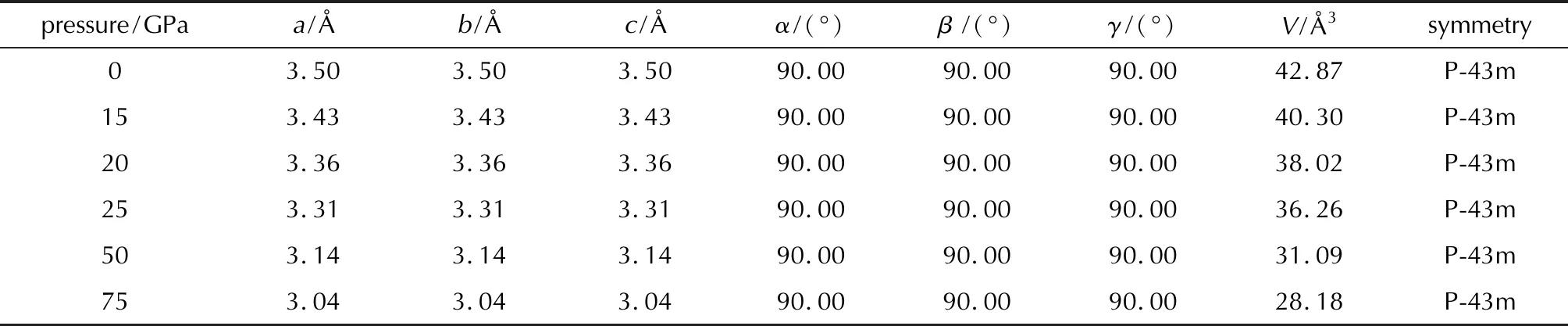
Table 4 Lattice parameters of P-43m-N4

Table 5 Lattice parameters of P4/m-LiN4
Table 6 Lattice parameters of Amm2-G/N4
The structure of P-43m-N4phase is shown in Fig.4(a),the most stable lattice parameters at 0GPa and 50GPa area=b=c=3.50Å anda=b=c=3.14Å,respectively.The bond length in the N4(Td)molecule is 1.46Å,indicating that it is still N—N single bond.The energy density of P-43m-N4phase is calculated to be 11.97kJ/g with respect to N2at ambient pressure,and the density of the crystal at 0GPa is 1.57g/cm3.The electron localization function (ELF)gives information of bonding between atoms.Fig.4(b)shows that there are localized lone-pair electrons at each corner of the N4tetrahedron.Such lone-pair electrons are repulsive to each other,and thus it takes higher pressure to stabilize the structure.
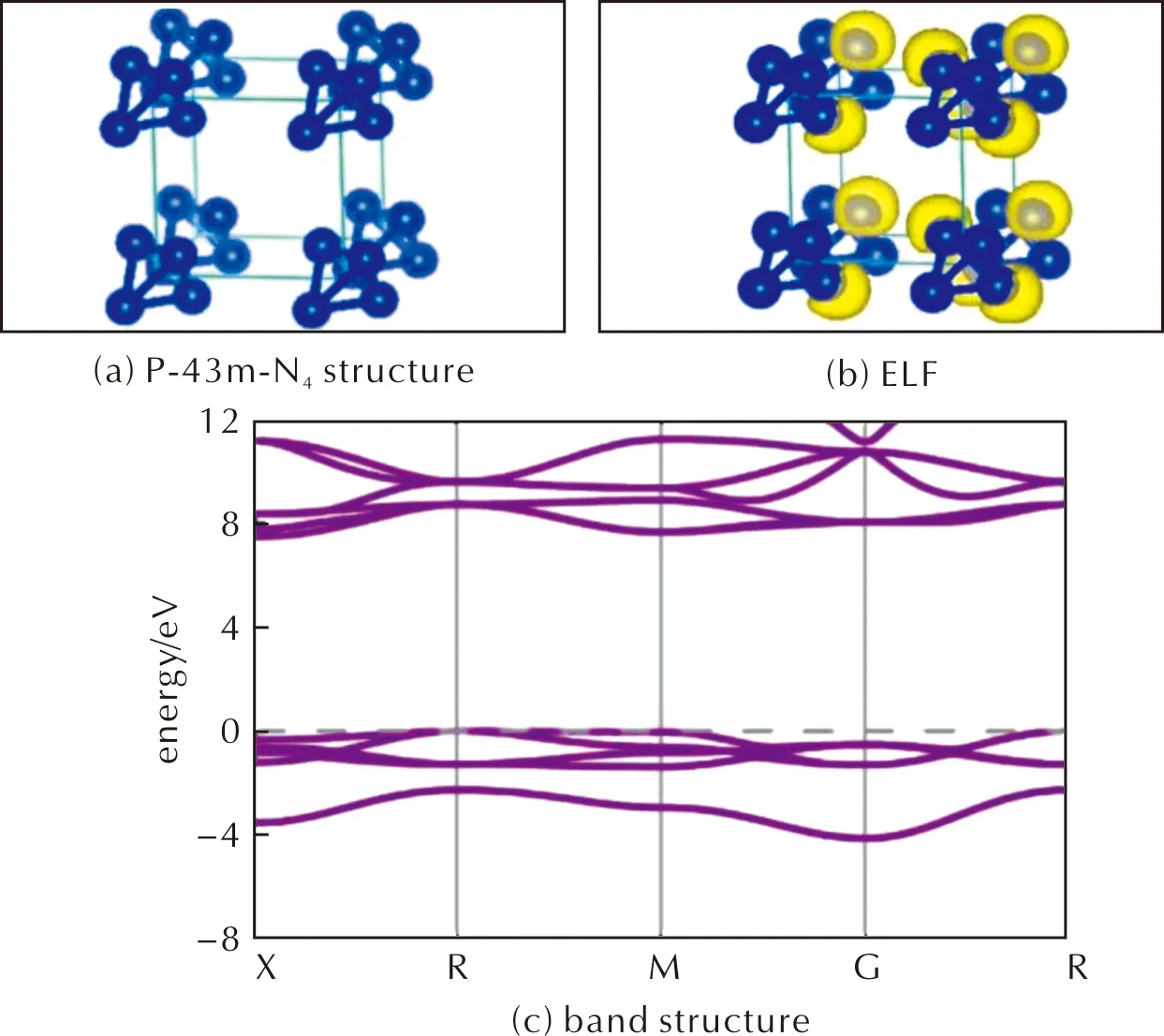

Fig.4 Molecular dynamics and thermodynamic properties of P-43m-N4. (c)was calculated under 0GPa. (d)was calculated under the NpT ensemble at 300K and 50GPa,the structure in the figure is supercell 3×3×3.
Fig.4(c)and Fig.5(a)shows that P-43m-N4is an insulator with a band-gap of 7.90eV,the valence band around Fermi-level is dominated by the N-2porbitals.To investigate the stability of the lattice,we have performed phonon band structure calculations,as shown in Fig.5(b)、(c)in Supplementary Materials.There are a few imaginary modes near theΓpoint,which are caused by the distortion of the lattice.We then perform AIMD simulations at 300K and 50GPa,the results indicate that N4molecules remain stable in the lattice,See Fig.4(d),while the lattice deviates from cubic slightly.The distorted lattice parameters after 10ps AIMD are shown in Table 7.At the same time,elastic constant was calculated to characterize the stability of the structure,but the structure does not satisfy the formula of elastic constant.P-43m-N4is Cubic crystal system,theC11,C44andC12are 22.36,-8.55,-2.09GPa,respectively.The structure is stable if the following calculation is satisfied:[C11>0,C44>0,C11>|C12|,(C11+2C12)>0].
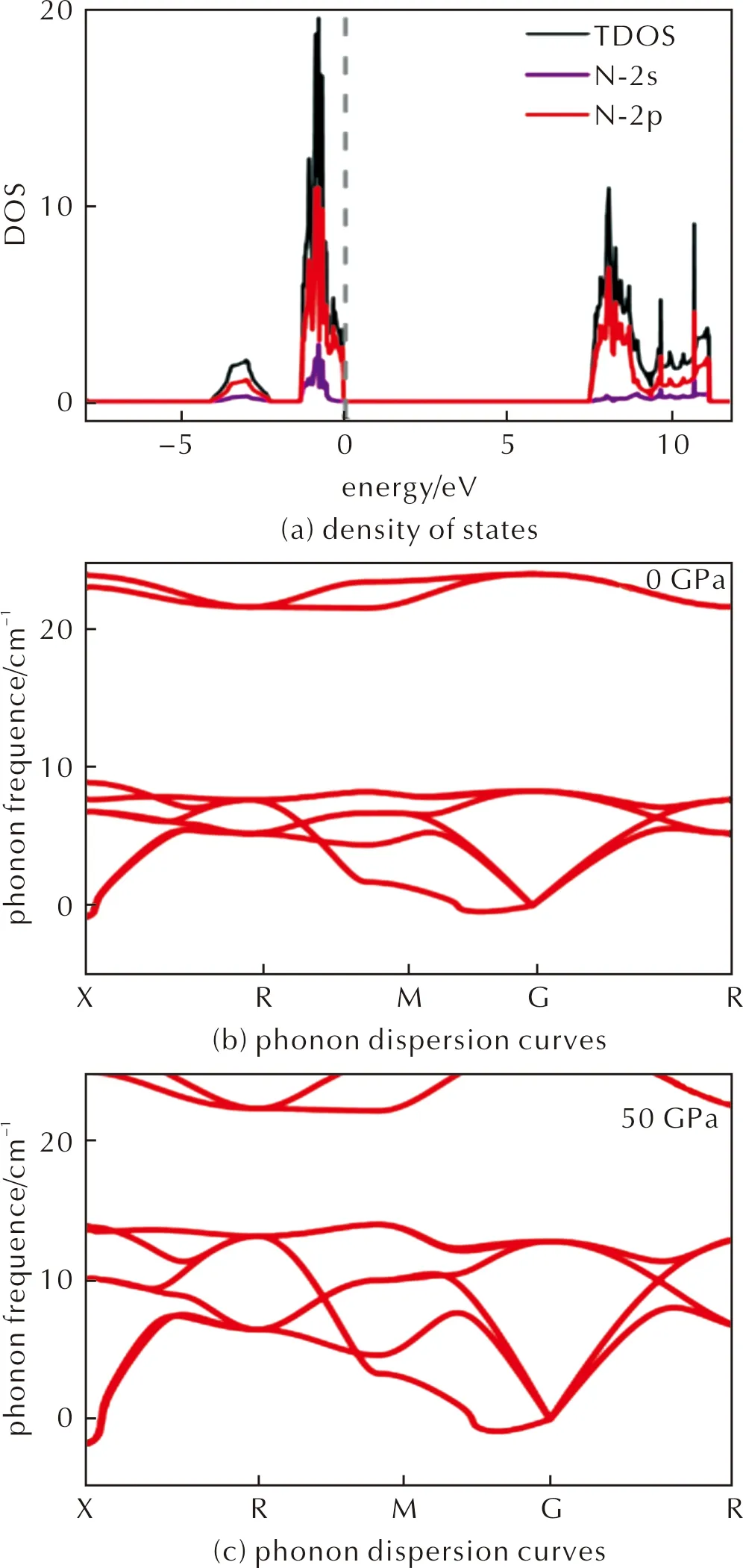
Fig.5 Thermodynamic properties of P-43m-N4. 3×3×3 supercell was used for calculation
Here we introduce the second crystal structure from the theoretical point of view is P4/m-LiN4,which is shown in Fig.6(a).The second crystal in this study is P4/m-LiN4phase shown in Fig.6(a),the distance between adjacent N atoms are all 1.35Å.
The lattice parameters of P4/m-LiN4structure under atmospheric pressure and 50GPa are calculated to bea=b=3.90Å,c=2.70Å anda=b=3.71Å,c=2.33Å,respectively.By decomposing to LiN3and N2at ambient pressure,P4/m-LiN4releases energy 4.68kJ/g.The ELF (Fig.6(b))indicates that there are localized lone-pair electrons between N atoms and Li atom so that they form a coordinate covalent bond.From Bader charge analysis,we see that Li atom transfers about 0.2 charges to each N atom.According to electronic structures calculations shown in Fig.6(c)and Fig.7(a)in Supplementary Materials,we can see that P4/m-LiN4is a metal phase,in which the conduction and valence bands near Fermi-level are mostly occupied by the N-2p orbitals.Next,we calculate the phonon band structures and the results are shown in Fig.7(b)and (c)for 0 and 50GPa.There are a few imaginary modes betweenMandΓpoints,which are mainly caused by the distortion of the lattice.Finally,AIMD simulations are performed at 300K and 50GPa,showing that although N4molecules remain,the lattice distorts and is no longer tetragonal lattice (See Fig.6(d)).The distorted lattice parameters after 10ps AIMD are also shown in Table 7.The elastic constant were also calculated,and the structure can satisfy the formula of elastic constant.P4/m-LiN4is Tetragonal,theC11,C33,C44,C66,C12,andC13are 533.53,489.62,60.59,3.61,45.42,23.74GPa,respectively.The structure is stable if the following calculation is satisfied:{[C11>0,C33>0,C44>0,C66>0,(C11-C12)>0,(C11+C33-2C13)]>0,[2(C11+C12)+C33+4C13]>0}.
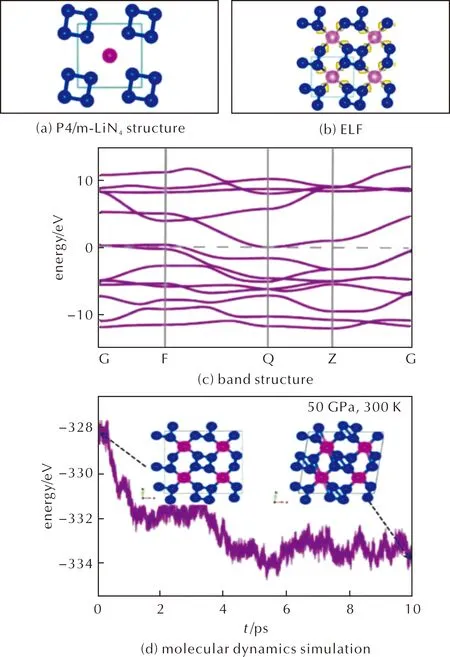
Fig.6 Molecular dynamics and thermodynamic properties of P4/m-LiN4 structure. (c)was calculated under 0GPa. (d)was calculated under the NpT ensemble at 50GPa and 300K,the structure here is supercell 2×2×4.
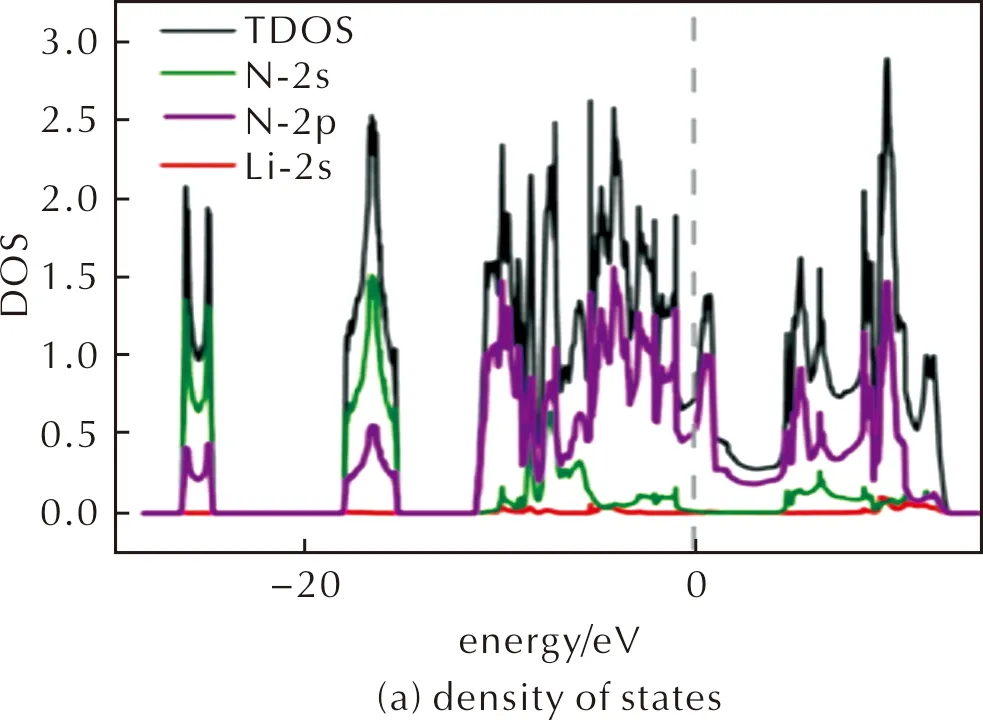

Fig.7 Thermodynamic properties of P4/m-LiN4. 2×2×2 supercell was used for calculation
The third crystal is Amm2-G/N4phase shown in Fig.8(a).The lattice parameters at ambient pressure and 50GPa area=b=4.30Å,c=6.50Å anda=b=4.06Å,c=5.49Å.The energy density is 7.35kJ/g at ambient pressure calculated with respect to graphene and N2.Again,we see lone-pair electrons around each corner of the N4(Td)molecule,as shown in the ELF of Fig.8(b).Band structures calculations shown in Fig.8(c)and Fig.9 in Supplementary Materials indicate that the crystal is a semimetal similar to graphene,and the valence and conduction bands are mostly composed of C-2porbitals.Mechanical stability depicted by phonon band structures are shown in Fig.9(b)and (c)for 0 and 50GPa in Supplementary Materials.There are also a few imaginary modes between theΓtoApoint,which are caused by the distortion of the lattice.


Fig.8 Molecular dynamics and thermodynamic properties of P Amm2-G/N4 structure. (c)was calculated under 0GPa. (d)was calculated under the NpT ensemble at 50GPa and 50K,the structure in the figure is supercell 2×2×2.
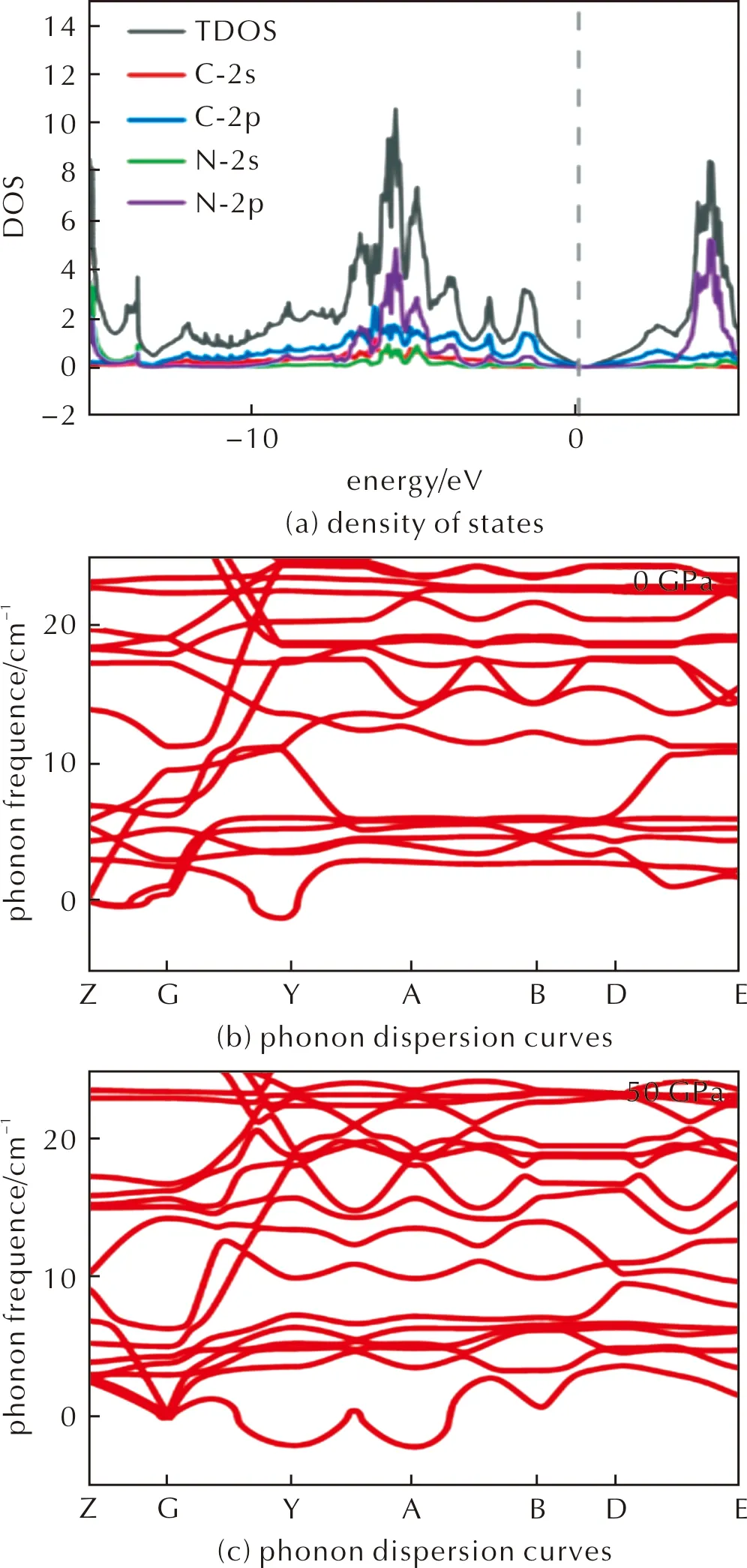
Fig.9 Thermodynamic properties of Amm2-G/N4. 2×2×2 supercell was used for the calculation
We next perform AIMD simulations at room temperature and 50GPa,N4(Td)decomposes into N2gas and thus decrease the temperature to 50K and repeat the calculation,as shown in Fig.8(d).At 50K and 50GPa,N4(Td)molecule is stable within 10ps,and the lattice is also distorted to release stress.The slightly changed lattice parameters are shown in Table 7.Elastic constants of the three crystals were also calculated.Amm2-G/N4is orthogonality,theC11,C22,C33,C44,C55,C66,C12,C13,C23are 690.92,690.91,297.26,279.10,408.65,-76.48,129.99,60.66,54.12GPa,respectively.The structure is stable if the following calculation is satisfied:{C11>0,C22>0,C33>0,C44>0,C55>0,C66>0,[C11+C22+C33+2(C12+C13+C23)]>0,(C11+C22-2C12)>0,(C11+C33-2C13)>0,(C22+C33-2C23)>0}.The results agree with the phonon spectra and AIMD simulations that distortion is necessary to stabilize the crystal under high pressure.
Finally,in order to guide further experimental research,we simulated the Raman spectra of the three crystals,as shown in Fig.10.

Fig.10 Three Raman spectra of the crystal structure.
For P-43m-N4,the Raman peaks near 1546.36cm-1and 924.054cm-1are mainly caused by the breathing and stretching modes of N—N bond,respectively.For P4/m-LiN4,an obvious Raman characteristic peak appears near 1074.521cm-1,which is mainly derived from the stretching mode of N—N bond.For Amm2-G/N4,there is a major peak at 1055.451cm-1,which is caused by the rotation of the N4molecule,and the peak near 1450cm-1is caused by the stretching mode of C—C bond and breathing mode of N—N bond.
3 Conclusion
(1)N4can be stabilized from perspectives of molecule and crystal.By adsorbing N4onto transition metal surfaces,it was found that the energy difference between N4and two N2molecules can be reduced from 6.48eV in gas phase to 3.24eV on metal surfaces.But N4will deform and lose the Tdsymmetry after interacting with the surfaces.From the crystal point of view,three crystals containing N4were proposed,which are P-43m-N4,P4/m-LiN4and Amm2-G/N4.
(2)We have calculated their energy densities,thermodynamic and kinetic stabilities,as well as their electronic properties.As the pressure goes up,the enthalpy differences between the crystals and their corresponding decomposition products decrease,showing that high pressure is an effective way to stabilize N4molecule.Pure N4crystal P-43m-N4has the highest energy density(11.97kJ/g)in this study,but due to the repulsive interaction between the lone-pair electrons in the N4,it requires the highest pressure to synthesis in the three crystals.In P4/m-LiN4,the lone-pair electrons in the N4ring form coordination bond with Li atoms,giving an energy density of 4.68kJ/g.Without such coordination bond in Amm2-G/N4,it needs temperature as low as 50K to be stable in our AIMD simulations.
(3)Our studies indicate that,through coordination bond between N4and metal atoms,polymerized N4can be synthesized as crystal at high pressure and stabilized at low temperatures.
