新型苯并咪唑衍生物的合成
2022-03-16何冠宁贾群坡
何冠宁, 袁 斌, 贾群坡, 吕 松
(广东工业大学 环境科学与工程学院,广东 广州 510006)
苯并咪唑其衍生物具有多种生物活性,如抗癌[1-4]、杀菌[5-7]、抗病毒[8-9]等。此外,苯并咪唑还可作为多种化工原料的中间体[10-11]和高效金属缓蚀剂[12-17]。在地沟油水解制备高级脂肪酸的过程中[18-19],会产生大量副产品甘油。然而,以地沟油水解副产物甘油为原料制备有机酸,进而合成出苯并咪唑类化合物的研究鲜有报道。
本文以苯甲醛甘油缩醛(1a)和对甲基苯甲醛甘油缩醛(1b)为原料,先采用次氯酸钠催化氧化法,制得了2-芳基-4-羧基-1,3-二氧五环中间体(2);2分别与邻苯二胺或N-甲基邻苯二胺反应,合成了新型苯并咪唑类化合物(3a~3d, Scheme 1)。收率为78.0%~80.5%,其结构经UV,1H NMR,13C NMR, IR和HR-MS(ESI)表征。采用正交实验设计,优化了中间体和苯并咪唑类化合物的合成条件。
1 实验部分
1.1 仪器与试剂
XT5B型熔点仪;Cary 100 UV-Vis型光谱仪;Bruker AVANCE III 600 MHz型核磁共振仪[(CD3)2SO为溶剂,TMS为内标];Thermo Fisher Nicolet 6700型红外光谱仪(KBr压片);Agilent 7890B-7000C型三重四极杆串联气质联用仪。
1a和1b按文献[20]方法合成;其余所用试剂均为分析纯。
1.2 合成
(1)2a和2b的合成(以2a为例)
将1a18.0 g(0.1 mol),适量2,2,6,6-四甲基哌啶氮氧化物(TEMPO),溴化钠和二氯甲烷30 mL加入三口烧瓶中,搅拌下,于0~10 ℃、pH 9~10的条件下,缓慢滴加14.5%次氯酸钠溶液72~86 g(0.14~0.17 mol),反应2~4 h(用KI-淀粉试纸检测次氯酸钠溶液用量是,直至试纸变蓝)。加入适量亚硫酸钠溶液,搅拌,分液,水相用10%盐酸调节pH至2~3,用甲基叔丁基醚(MTBE)(3×100 mL)萃取,合并有机相,依次用去离子水(2×50 mL)和饱和氯化钠溶液洗涤,旋蒸除溶得淡黄色黏稠液体2a6.44 g,收率35.8%, b.p.305~306 ℃; IRν: 3334(OH), 2914(CH2), 1705(C=O), 1614, 1503(苯环骨架), 1229(醚键), 752, 713(单取代苯)cm-1。
用1b替代1a,用类似的方法合成淡棕色黏稠液体2b 7.26 g,收率37.4 %, b.p.326~327 ℃; IRν: 3353(OH), 2955(CH2, CH3), 1746(C=O), 1198, 1121(醚键), 756(p-二取代苯)cm-1。
(2)3a~3d的合成通法
将一定量的2a或2b、邻苯二胺或N-甲基邻苯二胺盐酸盐和二甲苯20~25 mL加入三口烧瓶中,通氮气2 min,搅拌下缓慢加热至回流,酰化反应4~6 h。升温至190~210 ℃,环化反应10~14 h。冷却至室温,减压蒸出低沸点副产物,残余物用去离子水溶解,用等体积三氯甲烷萃取3次,合并有机相,旋蒸除溶,残余物用甲苯或丙酮重结晶,真空干燥得化合物3a~3d。
2-苯基-4-(2′-苯并咪唑基)-1,3-二氧五环(3a): 黑褐色固体2.15 g,收率81%, m.p.98~101 ℃; UV-Vis(EtOH)λ: 210, 240 nm;1H NMRδ: 8.23(s, 1H, NH), 7.78(s, 1H, CH), 7.63~7.45(m, 4H, C6H4), 7.31~7.20(m, 5H, C6H5),7.00(t,J=6.0 Hz, 1H, CH), 2.54(m, 2H, CH2);13C NMRδ: 151.90, 130.28, 129.47, 129.23, 127.93, 126.91, 126.50, 119.71, 111.92, 47.91; IRν: 3420.7, 3058.4, 1684.3, 1603.6, 1461.8, 1444.6, 1276.5, 763.4, 743.7, 698.5 cm-1; HR-MS(ESI)m/z: Calcd for C16H17N2O3{[M+H3O]+}285.19, found 285.14。
2-苯基-4-(1′-N-甲基-2′-苯并咪唑基)-1,3-二氧五环(3b): 黑色固体2.19 g,收率78.23%, m.p.105~107 ℃; UV-Vis(EtOH)λ: 220, 240 nm;1H NMRδ: 8.39(s, 1H, CH), 7.89~7.82(m, 4H, C6H4), 7.35~7.15(m, 5H, C6H5), 7.00(t,J=6.0 Hz, 1H, CH), 3.88(s, 2H, CH2), 3.89(s, 3H, CH3);13C NMRδ: 152.96, 137.26, 136.56, 136.20, 133.40, 132.12, 131.22 , 130.55, 130.55, 129.96, 129.54, 129.22, 127.39, 119.56, 111,39, 47.97, 32.37; IRν: 3057.0, 2885.1, 1603.6, 1516.3, 1469.7, 1442.8, 1277.2, 776.9, 745.1, 697.9 cm-1; HR-MS(ESI)m/z: Calcd for C17H19N2O3{[M+H3O]+}299.32, found 299.15。
2-(对甲苯基)-4-(2′-苯并咪唑基)-1,3-二氧五环(3c): 棕色固体2.31 g,收率80.47%, m.p.101~103 ℃; UV-Vis(EtOH)λ: 211, 239 nm;1H NMRδ: 8.13(d,J=6.0 Hz, 1H, NH), 7.74(d,J=6.0 Hz, 1H, CH), 7.63(m, 1H, CH), 7.41(d,J=6.0 Hz, 1H, CH), 7.30(d,J=12.0 Hz, 1H, CH), 7.0~7.24(m.4H, C6H4), 6.90(d,J=6.0 Hz, 2H, CH2), 2.36(m.1H, CH), 2.34(m.3H, CH3), 2.20(s.2H,CH2);13C NMRδ: 153.79, 151.87, 143.18, 139.98, 137.11, 136.35, 134.35, 129.96, 129.79, 129.38, 127.74, 126.91, 126.44, 122.59, 111.44, 47.74, 21,37 IRν: 3406.2(N—H), 3051.0, 2918.5, 1613.3, 1514.9, 1480.8, 1455.9, 1249.9, 823.1, 745.6 cm-1; HR-MS(ESI)m/z: Calcd for C17H20N2O4{[M+2H2O]+}315.32, found 315.18。
2-(对甲苯基)-4-(1′-N-甲基-2′-苯并咪唑基)-1,3-二氧五环(3d): 黑色固体2.34 g,收率79.58%, m.p.107~110 ℃, UV-Vis(EtOH)λ: 214, 241 nm;1H NMRδ: 7.75(d.J=12.0 Hz, 1H, CH), 7.71(d,J=6.0 Hz, 1H, CH), 7.63(d,J=12.0 Hz, 1H, CH), 7.59(d,J=12.0 Hz, 1H, CH), 7.42~7.06(m.4H, C6H4), 6.90(s, 1H, CH), 3.85(s, 3H, CH3), 2.39(s, 3H, CH3), 2.34(s, 1H, CH), 2.20(s, 2H, CH2);13C NMRδ: 153.41, 143.07, 142.43, 140.01, 137.11, 136.32, 134.33, 129.96, 129.78, 129.38, 127.62, 126.99, 126.44, 119.12(CH), 111.03(CH), 47.74, 32.11, 21.41; IRν: 3048.9, 2917.8, 1612.7, 1513.6, 1458.2, 1249.3, 1183.0, 822.8, 747.6 c m-1; HR-MS(ESI)m/z: Calcd for C18H21N2O3{[M+H3O]+}313.35, found 313.17。
2 结果与讨论
2.1 合成
表1为2a的正交实验结果。由表1的极差分析结果可知,各因素对化合物2a合成的影响顺序为:NaBr量>TEMPO量>氧化温度>氧化时间。综合考虑各个因素的影响,可确定该化合物的最佳合成条件为:催化剂TEMPO用量为苯甲醛甘油缩醛原料的2%,氧化温度为5 ℃,氧化时间为6h, NaBr用量为2 g。

表1 合成2a的正交实验
以化合物(3a)的合成正交实验为例,说明4种化合物合成最佳工艺条件的选择。化合物3a的正交合成实验结果见表2。通过类似的正交实验设计,分别优选出其它3种新物质的合成工艺条件,最佳合成实验结果见表3所示。
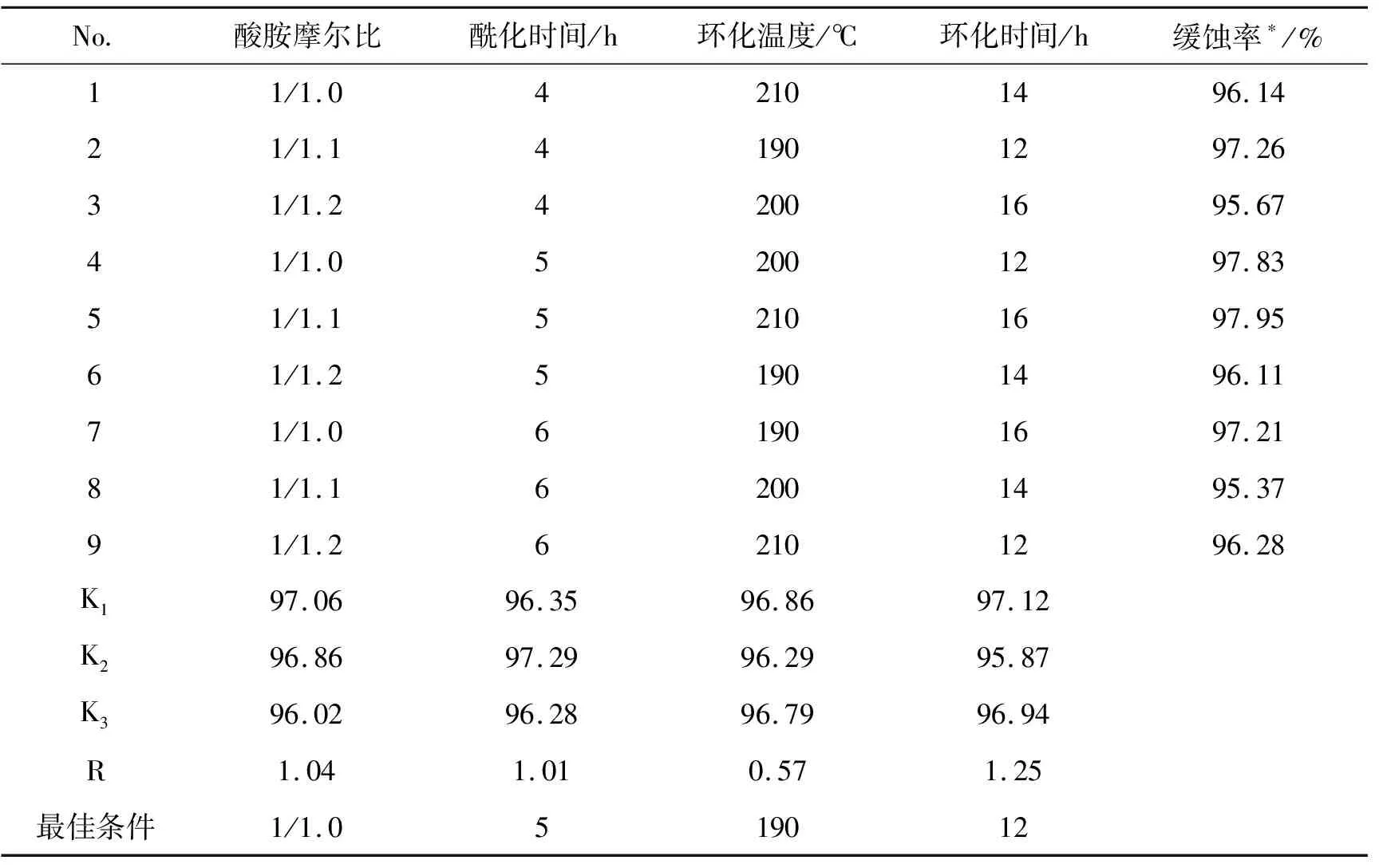
表2 合成3a的正交实验
从表2可见,苯并咪唑化合物3a合成的影响因素次序为:环化时间>酸胺原料摩尔比>酰化时间>环化温度,这表明此类化合物合成必须保证足够的环化反应时间。
从表3可见,4种苯并咪唑类化合物的最优合成条件为:酸胺摩尔比为1/1.0,酰化时间5~6 h,环化温度190~210 ℃,环化时间12~14 h。这表明4种新化合物的合成条件基本相同。

表3 3a~3d的最佳合成条件
2.2 表征
(1)IR
由IR谱图(图略)可知,与原料1a的红外谱图对比,2a在1741 cm-1处出现了羧基的C=O特征振动吸收峰,证明中间产物2a已被合成。
3a和3c在3396~3420 cm-1出现了N—H伸缩振动吸收峰,3a~3d在2917~3038 cm-1出现了饱和C—H伸缩振动吸收峰,在1684~1444 cm-1出现了咪唑环和苯环骨架的特征吸收峰,3a和3b在745~697 cm-1出现了单取代苯的特征吸收峰,3c和3d在747, 823 cm-1附近出现了二取代苯的特征吸收峰。上述的特征吸收峰与苯并咪唑衍生物的官能团吸收峰相符,证明4种苯并咪唑衍生物均已被成功合成[21]。
(2)UV-Vis
图1为3a~3d的UV-Vis谱图。由图1可知,210~220 nm处均出现了苯并咪唑环结构的特征吸收峰[22],这与邻苯二胺的紫外吸收特征波长254 nm和苯环的紫外吸收特征波长180 nm及200 nm均有显著区别,由此证明了苯并咪唑环已被成功合成。

λ/nm
(3)1H NMR
由1H NMR谱图(图略)可知,3b除了在δ3.89处出现了甲基吸收峰外,其余吸收峰与3a的特征峰基本一致;3d除了在δ3.85处出现了甲基吸收峰以外,其余吸收峰与3c的特征峰 基本一致。
以苯甲醛甘油缩醛和对甲基苯甲醛甘油缩醛为原料,合成了4种新型的苯并咪唑类化合物,收率78.0%~80.5%。该方法具有操作简单、收率较高等优点。
