骨代谢相关因子参与动脉粥样硬化的研究进展
2022-03-13孙婷婷
孙婷婷 王 凯
心脑血管疾病(尤其冠心病)和骨质疏松症是老年人群普遍存在的健康问题,随着人口老龄化的加剧,该类疾病严重影响患者日常生活能力、生活质量,甚至影响患者预期寿命。随着临床数据积累,越来越多证据表明,心血管疾病患者中骨量低下、骨质疏松症及脆性骨折发病率升高。同时,在骨质疏松患病人群中,心血管疾病风险亦有增高趋势。Persy等[1]将这种相关性解释为“骨-血管钙化悖论”, 定义为骨丢失和血管钙化的一致性,描述血管内异位钙化常伴随着骨密度降低或骨转换紊乱的现象。其病理生理学机制包括:血管平滑肌细胞(vascular smooth muscle cell, VSMC)增殖和迁移、氧化应激、内皮损伤、血管钙化、炎症反应等多个途径。绝经后状态、运动缺乏、吸烟、高龄等是两者发病的共同危险因素,并在两者间相互影响。本文主要总结骨代谢相关因子对动脉粥样硬化的影响(相关机制及其结局见表1),为临床治疗骨质疏松和心脑血管疾病提供新治疗思路。
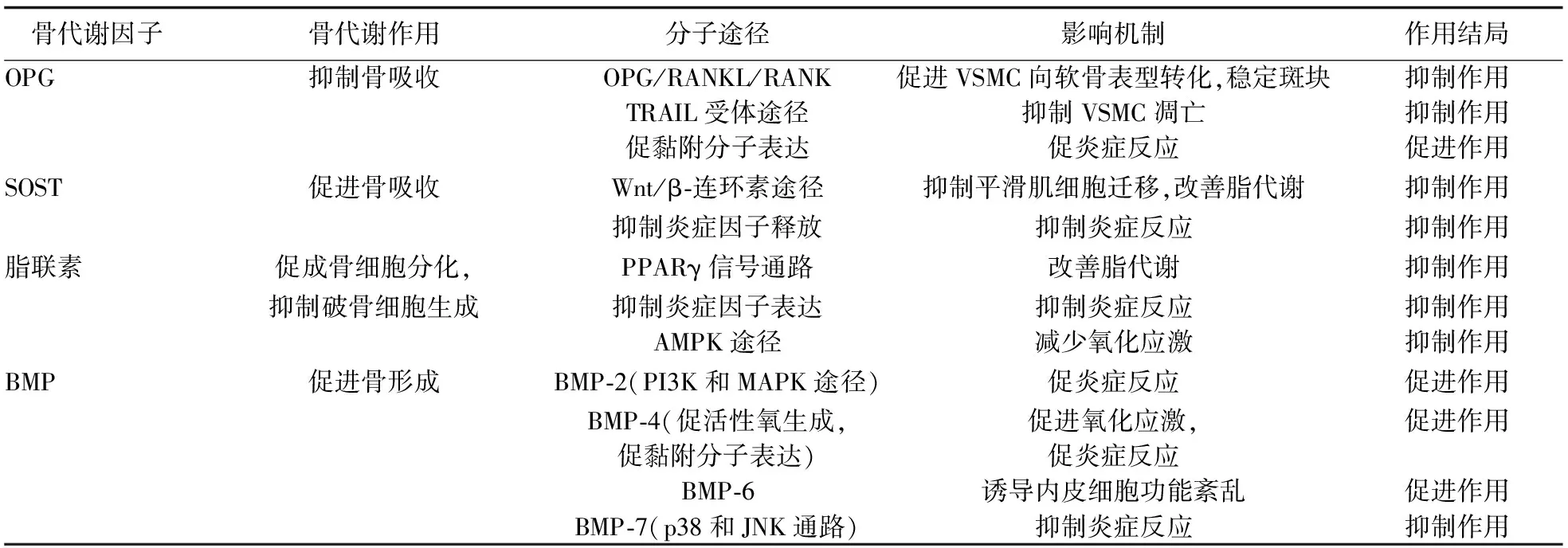
表1 骨代谢相关因子对动脉粥样硬化的影响机制及其结局
1 OPG
OPG是由成纤维细胞分泌的TNF受体超家族诱导的可溶性细胞因子,通过抑制破骨细胞的生成参与骨代谢。核因子κB受体活化因子(receptor activator of nuclear factor-kappa B,RANK)和核因子κB受体活化因子配体(receptor activator for nuclear factor-κ B ligand,RANKL)是TNF受体超家族的一对受体和配体,两者结合后诱导破骨细胞分化和激活成熟破骨细胞来促进骨吸收,是骨代谢的关键途径,OPG作为可溶性诱饵受体,与RANK竞争结合RANKL,抑制RANKL与RANK相互作用,从而抑制破骨细胞生成[2]。OPG除在成骨细胞中表达外,在内皮细胞、VSMC和巨噬细胞中均有表达[3]。OPG参与动脉粥样硬化进展主要与其RANKL/RANK、TNF相关凋亡配体(TNF related apoptosis inducing ligand, TRAIL)信号转导通路及炎症反应相关。①OPG/RANKL/RANK通路作用。RANKL显著提高VSMC表达的基质金属蛋白酶(matrix metalloproteinase,MMP)活性[4],MMP在动脉粥样硬化斑块的基质降解中起关键作用,可使斑块易于破裂,从而发挥血管损伤作用。OPG通过抑制RANKL与RANK的结合来中和RANKL对MMP活性的诱导作用,从而稳定粥样硬化斑块。另外,RANK/RANKL水平上调,可以促进正常VSMC向软骨细胞样表型转化引起血管钙化,OPG竞争性与RANKL结合可保护VSMC表型分化[5]。②TRAIL受体主要包括5种,其中TRAIL R1、TRAIL R2为死亡受体,胞内具有死亡功能区,与TRAIL 特异性结合可通过死亡结构域变构激发和转导死亡信号,诱导细胞死亡。TRAIL在脂肪细胞、内皮细胞、VSMC内表达诱导局部细胞凋亡,促进稳定斑块向易破裂斑块转化[6],发挥促动脉粥样硬化作用。OPG作为诱导型受体,通过与TRAIL配体结合,抑制TRAIL与死亡受体结合诱导的VSMC凋亡,从而影响血管壁的代谢过程[7]。除此之外,TRAIL还可通过其抗炎特性和促进局部NO的产生而对内皮细胞产生保护作用[8],该作用是否与OPG相关尚无定论。③OPG影响炎症反应。血管内皮细胞在TNF-α、IL-1等炎症因子作用下释放OPG,OPG诱导内皮细胞间黏附分子(如血管黏附分子-1和E-选择素)表达,促进白细胞黏附,导致内皮细胞早期功能障碍,促使动脉粥样硬化发生[9]。研究[9]显示,高血浆OPG水平可以通过增强炎症反应、促进炎症细胞在斑块中的浸润、增加血管生成素-Ⅱ(angiopoietin Ⅱ,Ang-Ⅱ)的表达和增强内皮细胞的黏附等,促进缺血性卒中的发生并加重卒中的严重程度。尽管目前尚不能确定OPG在动脉粥样硬化进展中的最终作用,但仍有不少学者探讨了OPG作为评价动脉粥样硬化的生物学标志物的价值[10-11]。
2 SOST
SOST是一种主要由骨细胞分泌的糖蛋白,是Wnt介导的成骨细胞活化和骨形成的抑制剂,参与骨吸收和炎症性骨丢失[12],为代谢性骨病治疗策略的进展开辟了一个新的领域。SOST不仅在骨细胞内表达,在动脉硬化血管平滑肌及钙化的主动脉和颈动脉斑块内也过度表达。SOST对动脉粥样硬化的影响,主要是其作为Wnt信号转导通路抑制剂,抑制Wnt在血管内皮细胞及VSMC中的作用。β-连环素(β-catenin)是Wnt通路的核心成分,Wnt配体通过与Frizzled受体和脂蛋白受体相关蛋白(lipoprotein receptor-related protein,LRP)5、LRP6共受体结合稳定连环素的结构,使细胞质中β-连环素的破坏复合物失活,促进其转移到细胞核内参与基因转录与调控。Wnt/β-连环素途径在动脉粥样硬化中表达上调,并参与从内皮功能障碍到血管钙化的不同阶段[13]。其具体作用如下:①当内皮细胞受到损伤,血管内炎症和血栓前基因被诱导表达,导致炎症细胞向内皮下间隙浸润,使循环中的低密度脂蛋白(low density lipoprotein, LDL)水平增高;此时出现LRP5受体过表达,促进单核细胞向巨噬细胞转化,吞噬LDL形成泡沫细胞[14];同时,内皮细胞损伤将激活Wnt信号通路,导致β-连环素表达增加,进一步加重炎症因子的浸润,促进动脉斑块的形成[15]。② Wnt信号通过调节MMP的表达调控平滑肌细胞的增殖、迁移和凋亡,以及向成骨细胞表型转化[16],导致平滑肌细胞向内膜下迁移致管腔狭窄及血管钙化,并促进动脉管壁硬化。③ Wnt5a通路是非经典Wnt信号代表,与经典Wnt信号作用相互竞争,研究显示Wnt5a通道及其受体、清道夫受体在巨噬细胞及泡沫细胞内表达上调,加速斑块发生不稳定及破裂的趋势[17]。
SOST通过抑制糖原合成酶激酶3(glycogen synthase kinase 3,GSK-3)磷酸化,抑制GSK-3对β-连环素及Wnt信号通路激活[18],延缓动脉粥样硬化进展。在动脉硬化或缺血事件中,SOST作为Wnt信号抑制剂的表达上调,可能是抑制Wnt途径激活的应答反应[19]。除了对Wnt信号的抑制作用,SOST还可抑制Ang-Ⅱ灌注诱导的小鼠细胞内炎症因子[包括TNF、IL-6、单核细胞趋化蛋白-1(monocyte chemoattractant protein,MCP-1)、IFN-γ]释放增加,起到抗动脉粥样硬化的作用[18]。研究[20]显示,老年稳定型冠心病PCI术后,高SOST水平者主要心脑血管病不良事件发生率低于低SOST水平者,远期生存率高于低SOST水平者。He等[21]研究发现,SOST作为大动脉缺血卒中标志物,其最佳阈值为125.8 ng/L,灵敏度为68.6%,特异度为77.4%。
3 脂联素(adiponectin)
脂联素是脂肪因子的一种成分,由脂肪细胞及成骨细胞分泌,并且在成骨细胞及破骨细胞中均有表达。研究结果表明,脂联素直接影响成骨细胞的增殖和分化,并抑制TNF-α和RANKL诱导的破骨细胞生成。脂联素具有提高胰岛素敏感性和抗炎作用,能刺激骨钙素的表达和成骨细胞的分化,并能抑制骨髓中脂肪的加速生成[22]。
脂联素参与动脉粥样硬化进展主要表现为对脂代谢、VSMC增殖和迁移、炎症反应的影响。脂联素通过激活肝X受体α和过氧化物酶体增殖激活受体γ(peroxisome proliferators-activated receptors,PPARγ)信号通路增强ATP结合盒转运体A1(ATP-binding cassette transporters A1,ABCA1)的表达促进高密度脂蛋白水平升高,继而通过增加肝内载脂蛋白apoA-Ⅰ和ABCA1的产生,反向转运胆固醇增高HDL-C水平[23]。脂联素可以下调血清载脂蛋白CⅢ水平,后者的下调减少了对脂蛋白酯酶的抑制作用,使脂蛋白酯酶活性增加;脂联素也可直接诱导骨骼肌及脂肪组织脂蛋白酯酶的表达[24-25],分解富含TG的脂蛋白。脂联素通过抑制前脂肪细胞因子-1表达及增加PPARγ表达,作用于心外膜脂肪组织,抑制了MCP-1、IL-6、IL-8和TNF-α等炎症因子的表达,改善前脂肪细胞向成熟脂肪细胞分化,加速内脏脂肪组织代谢,维持正常的脂质水平[26]。脂联素通过抑制血小板源性生长因子、碱性成纤维细胞生长因子和肝素结合表皮生长因子活性,阻止VSMC的增殖和迁移[24,27],抑制动脉粥样硬化发生的早期步骤。脂联素通过与脂联素受体1结合,激活AMP活化蛋白激酶(AMP-activated protein kinase,AMPK)信号通路,促进内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS)的表达,增加NO合成[28],改善血管内皮功能及冠状动脉的供血。动物实验研究[29]亦显示,补充外源性脂联素通过增加eNOS mRNA的表达及超氧化物歧化酶的活性,减少氧化应激反应,可以减小动脉粥样硬化斑块面积。
4 BMP
BMP是TGF-β超家族成员之一, 最初被发现是由于其能诱导异位组织骨化和软骨形成。有研究[30]显示,重组人BMP-7已被临床用于诱导骨折周围间充质干细胞分化为软骨细胞、破骨细胞,通过钙沉积形成新骨,以及促进骨折修复。在不同的BMP中,BMP-2、BMP-4、BMP-6和BMP-7在血管生物学中发挥着重要作用。在慢性炎症条件下,血管内皮及平滑肌细胞分泌更多TNF-α,以旁分泌的方式诱导内皮细胞BMP-2的表达,并刺激含BMP-2的内皮微粒释放[31],在血管损伤部位,内皮微粒与血管平滑肌结合,促进VSMC的成骨转化,从而促进血管钙化[30]。BMP-2通过磷脂酰肌醇3激酶(phosphoinositide 3-kinase,PI3K)和丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)途径诱导单核细胞迁移,并促进单核细胞向促炎性M1巨噬细胞转化,释放TNF-α、IL-1β等炎症因子,增强内皮炎症反应,增加对单核细胞的黏附性[31],发挥促动脉粥样硬化作用。Csiszar 等[32]的研究结果显示,BMP-4能上调冠状动脉内皮细胞NADPH氧化酶的表达,增加活性氧生成,提高氧化应激水平;另外BMP-4增加血管内皮黏附分子的表达,增强细胞对内皮的黏附性,促进动脉粥样硬化的进展。Buyukterzi 等[33]研究发现,ACS患者的血清BMP-4水平较正常冠状动脉组升高,认为BMP-4可能是冠心病发展的独立危险因素。BMP-6和氧化低密度脂蛋白单独并且协同地调节成骨细胞分化的关键转录调节因子和骨桥蛋白的表达[34]。血管损伤时,骨桥蛋白在血管内表达,凝血酶等炎症介质将其诱导裂解为碎片,促进T细胞和单核-巨噬细胞向内皮细胞及平滑肌细胞趋化,使eNOS表达减少致NO合成减少,导致内皮细胞功能紊乱[35],从而促进动脉粥样硬化形成。Singla等[36]研究显示,在ApoE-/-小鼠模型行部分左侧颈动脉结扎术后,外源补充BMP-7可改善小鼠颈动脉粥样硬化程度,同时小鼠体内抑制炎症反应因子(IL-6、TNF-α、MCP-1)水平上调,与BMP-2不同的是,小鼠颈动脉处单核细胞浸润程度较轻,单核细胞向M2巨噬细胞转化增加,抗炎症因子(IL-10、 IL-1受体拮抗剂)水平上调;BMP-7发挥上述作用,可能与降低炎症p38和c-Jun氨基端蛋白激酶(c-Jun N-terminal kinase,JNK)通路的激活相关[30]。
综上所述,骨代谢相关因子在骨质疏松过程中参与了骨形成或骨吸收,又通过多个途径影响动脉粥样硬化各个阶段,体现了骨质疏松与动脉粥样硬化之间的相互联系。尽管这些相关因子动脉在粥样硬化进展中的作用不同,但都可能为今后研究心脑血管疾病治疗新靶点提供临床依据。
