甲基丙二酸血症的临床表型及基因特征分析
2022-03-11赵婉晴张亚男魏晨曦白欣立
赵婉晴,张亚男,魏晨曦,白欣立
(河北医科大学第二医院 儿科,河北 石家庄 050000)
甲基丙二酸血症(methylmalonic acidemia,MMA)是一种常染色体隐性遗传代谢病,由甲基丙二酰辅酶A变位酶(methylmalonyl-CoA mutase, MCM)及其辅因子钴胺素(维生素B12)代谢障碍引起。患者体内甲基丙二酸及其代谢产物异常蓄积,导致神经、肾脏、血液等多个系统受损[1-2]。
MMA可发病于任何年龄,大部分患者为早发型,在生后数分钟至数周内出现临床症状,主要表现为反复呕吐、嗜睡、昏迷、脱水、血氨升高等,病死率高,预后较差,长期并发症主要表现为神经和肾脏的损伤[3]。患者基因突变类型不同,发病时间、病情严重程度、临床表现也千变万化[4-7]。根据血清同型半胱氨酸水平是否升高,MMA分为合并型和单纯型。单纯型仅表现为MMA、不伴随血清同型半胱氨酸水平的升高,包括:mut型、cblA型、cblB型及cblD型[8]。其中,mut型是最常见的单纯型MMA,为甲基丙二酰辅酶A变位酶基因(methylmalonyl-CoA mutase gene, MUT)基因突变所致,以反复出现的难以纠正的代谢紊乱为主要表现。本研究对河北医科大学第二医院儿科内分泌遗传代谢诊室诊治的18例mut型MMA患儿的临床资料进行了回顾性分析,并对相关文献进行复习,为mut型MMA的临床诊断及治疗提供依据。
1 资料与方法
1.1病例选择 收集2015年12月至2020年6月于河北医科大学第二医院儿科内分泌遗传代谢诊室诊治的mut型MMA患儿18例,男11例,女7例。其中5例通过新生儿筛查确诊,其余13例为发病后确诊。所有患儿分别来自于不同的家庭,父母均非近亲结婚,母亲怀孕史及患儿出生史均无异常表现。本项研究经过河北医科大学第二医院伦理委员会批准,所有检查均获得了患儿及家属同意。
1.2方法 回顾性分析患儿的临床资料,包括:①临床表现:抽搐、呕吐、嗜睡、喂养困难等;②一般检查:血氨、同型半胱氨酸、肝功能、肾功能、β2微球蛋白、胱抑素C;③基因检测:Sanger测序或高通量测序;④氨基酸及酰基肉碱检测:丙酰肉碱(propionyl carnitine,C3)、乙酰肉碱(acetyl carnitine,C2)、C3/C2、游离肉碱(free carnitine,C0)、甲硫氨酸(methionine, Met);⑤尿有机酸检测:甲基丙二酸、甲基枸橼酸-4及3-羟基丙酸。其中氨基酸、酰基肉碱检测采用气相色谱-质谱联用技术检测,尿有机酸测定采用气相色谱-质谱联用技术检测。
2 结 果
2.1临床表现及一般检查 18例mut型MMA患儿中,男11例,女7例。5例经新生儿遗传代谢病串联质谱筛查确诊,其余患儿就诊年龄为3天至4岁。13例非新生儿筛查确诊的患儿中,1岁内发病9例,为早发型,表现为喂养困难、呕吐、嗜睡、精神不佳、生长发育落后等;1岁后发病4例,为晚发型,多表现为呕吐、抽搐、精神不佳等。患儿临床表现多样化、无特异性,呕吐7例,精神不佳5例,嗜睡4例,生长发育落后于同龄正常儿童4例,喂养困难3例,呼吸深大3例,抽搐2例,发热2例,恶心、意识不清、腹泻各1例。18例mut型MMA患儿中,同型半胱氨酸均正常(缺失数据者除外),肝功能均无明显异常。大部分患儿发病时血氨水平有所升高,且造成不同程度的肾损伤。见表1。
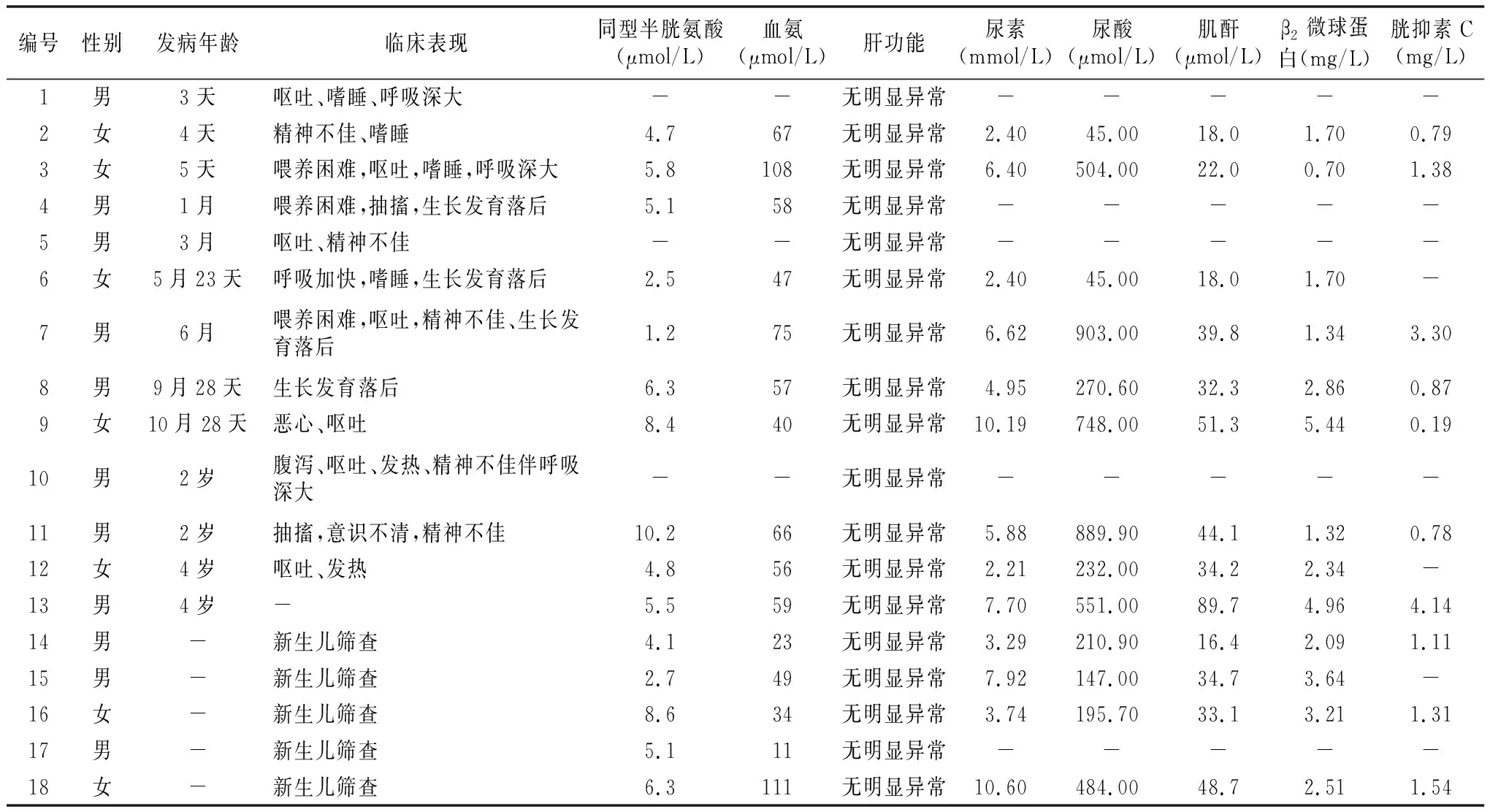
表1 18例mut型MMA患儿临床表现及一般检查
2.2血尿代谢及基因检测 18例患儿普遍出现血C3升高,C3/C2升高;尿有机酸中,甲基丙二酸显著升高,甲基枸橼酸-4有不同程度的升高。18例患儿均为MUT基因突变,复合杂合变异16例,共检测到34个突变位点,累计20种,突变主要集中在Exon3,共计10次,突变频率为29.4%(10/34);Exon6共计5次,突变频率为14.7%(5/34);Exon2共计4次,突变频率为11.8%(4/34)。基因突变类型多样化,包括10种错义突变,具体为:c.1808G>A、c.323G>A、c.599T>C、c.1106G>A、c.424A>G、c.1663G>A、c.1280G>A、c.947A>C、c.914T>C、c.1295A>C;无义突变5种:c.1741C>T、c.2179C>T、c.1630-1631delinsTA、c.2009G>T、c.2080C>T;移码突变2种:c.755dupA、c.920_923del;整码突变1种:c.1233_1235delCAT;剪切位点突变1种:c.1677-1G>A;插入突变1种:c.729_730insTT;起始密码子突变1种:c.556A>G;内含子突变1种:c.1084-10T>C。突变次数最多的位点为c.729_730insTT,突变频率为14.7%(5/34),其次为c.323G>A 、c.1106G>A,突变频率分别为11.8%(4/34)、8.8%(3/34)。见表2。

表2 18例mut患儿基因检测与血尿代谢结果
3 讨 论
MMA是以有机酸代谢紊乱为特点的罕见遗传病,由MCM及其辅因子钴胺素(维生素B12)代谢障碍引起,主要遗传方式为常染色体隐性遗传,甲基丙二酸和代谢物的异常蓄积导致患儿出现各种各样的临床症状。MCM完全或者部分活性丧失为单纯型MMA(即mut型),表现为MCM的辅因子钴胺素(cb1A、cb1B、cb1D-MMA、cb1H)的转运及合成障碍或缺乏MCM[2,9]。其中MUT基因突变是国内外最常见的单纯型MMA突变类型,据有关文献报道,目前MUT基因突变类型约250多种[2,10]。
单纯型MMA的临床表现为非特异性。本研究中,18例mut型MMA患儿临床表现较为复杂,基因类型不同的患儿临床表现也存在相似之处,多表现为呕吐、嗜睡、精神不佳、喂养困难等,严重者甚至出现抽搐、呼吸困难、意识障碍等,与既往文献报道一致[10]。当患儿出现多系统表现时,临床上应怀疑MMA的可能性。本研究中,5例于新生儿筛查时确诊,占比27.8%,高于国内其他文献报道比例[10-11]。新生儿筛查检出率的提高说明,我国对于新生儿早期筛查越来越重视,新生儿筛查对MMA的早诊断、早治疗至关重要。目前,我国有越来越多的地区开展了新生儿串联质谱筛查项目,有利于准确、及时地诊断多种疾病。本研究中,早发型9例,占比69.2%,为主要发病类型,最主要的临床表现为嗜睡、呕吐、喂养困难、精神不佳,可合并发热、抽搐、呼吸困难、生长发育落后等表现,但上述症状在婴幼儿期特异性较差[12]。因此,当患儿存在相关家族史或出现不明原因发热、抽搐、呼吸困难、生长发育落后等症状时,应考虑到MMA的可能性,及时对患儿进行血尿代谢的筛查及基因检测以排除诊断。
基因检测技术的不断提高为MMA的诊断提供了帮助,使越来越多的患儿得以早期诊断,并在临床症状出现前得到及时治疗。随着二代测序技术的发展,基因检测是确诊MMA及其分型最可靠的方法。MUT基因位于染色体6p12.3区域,共包含13个外显子,全长约35 kb[12]。MUT基因在不同种族中存在差异,在日本主要为c.349G>T(p.E117X)、c.385+5G>A(IVS2+5G>A)、c.1106G>A(p.R369H)、c.1481T>A(p.L494X)、c.2179C>T(p.R727X),在印度主要为c.1863A>T(p.K621N)、c.1943G>A(p.G648D)、c.1889G>A(p.G630E);在西班牙人中以c.322C>T(p.R108C)最常见;在黑色人种中以c.2150G>T(p.G717V)最常见;在我国,主要报道位点为c.729_730insTT(p.D244Lfs*39)、c.1106G>A(p.R369H)等[11]。本研究中,对18例mut型MMA患儿进行Sanger测序或高通量测序,其父母均为正常无表型,采用Sanger验证,患儿基因突变均为杂合,共检测到20种突变位点:c.323G>A、c.599T>C、c.1106G>A、c.424A>G、c.1663G>A、c.1280G>A、c.947A>C、c.914T>C、c.1295A>C、c.1741C>T、c.2179C>T、c.1630-1631delinsTA、c.2009G>T、c.2080C>14T、c.755dupA、c.920_923del、c.1233_1235delCAT、c.1677_1G>A、c.729_730insTT、c.556A>G。其中,最常见的突变类型分别为:c.729_730insTT(p.D244fs)5例,占比为14.7%;c.323G>A(p.R108H)4例,占比为11.8%;c.1106G>A(p.R369H)3例,占比为8.8%,与既往报道存在一定的差异,考虑可能与样本量偏小有关[11,13]。本研究中,基因突变类型多样化,主要表现为错义突变,共19个,占比55.9%。变异位点主要位于 N-末端结构域的第2、3、6外显子,通过引起蛋白质二级结构的改变,从而影响 MCM 的活性,与既往文献报道一致[14-15]。有研究收集了来自中国多家医院c.1663G>A (p.A555T)的复合杂合子患者30例,详细描述了它们的临床特征和生化指标,并与无此位点变异的患者进行了比较,结果显示,存在c.1663G>A (p.A555T)位点突变的患者发病较晚,临床表现较轻,生化异常较轻,对维生素B12的反应性较好,发病率较低,代谢控制较容易,预后较好[16]。本研究中,2例c.1663G>A突变位点携带者均为新生儿筛查发现,治疗过程中患儿对维生素B12反应较好,代谢控制较容易,与Liang等[16]研究报道一致。
本研究中, C3升高14例、C3/C2升高16例,尿甲基丙二酸升高18例,甲基枸橼酸-4(一)升高10例,甲基枸橼酸-4(二)升高9例, 3-羟基丙酸升高8例,与既往文献报道一致,可基本代表此型患儿的血尿代谢特征[17-19]。
MMA患儿具有临床表现复杂及早发严重的倾向,临床工作中容易出现误诊、漏诊。为减少这种情况的发生,对于以反复呕吐、嗜睡、精神不佳等为主诉就诊的患儿,应警惕MMA的可能性。尤其对于存在相关家族史或出现不明原因发热、抽搐、呼吸困难、生长发育落后的患儿,应及时进行血尿代谢筛查及基因检测并综合分析,达到早诊断、早治疗。该病治疗遵循个体化原则,可通过饮食、药物、手术等方式干预疾病进展,改善患者症状。维生素B12治疗有效的患儿饮食无需特殊限制,可主要通过补充维生素B12,其次通过口服左卡尼汀、甜菜碱、亚叶酸钙可明显改善症状;维生素B12治疗无效或部分有效的患儿主要通过饮食治疗,其次通过补充左卡尼汀、特殊奶粉、限制摄入蛋白质中氨基酸的含量等方式进行改善[20]。最后,对于饮食和药物控制不良的患儿可采取肝脏移植的手段进行治疗。有研究对9例接受肝脏移植的MMA肺动脉高压患儿及2例接受肝脏移植和肾脏移植的MMA肺动脉高压患儿进行了回顾性分析,结果表明肝脏移植可明显改善MMA患儿代谢,提高患儿5年生存率[21]。MMA可出现多种并发症,主要包括智力障碍、肾小管间质性肾炎伴进行性肾功能衰竭、“代谢性中风”(急性和慢性基底神经节损伤)导致瘫痪性运动障碍,伴有舞蹈症、肌张力障碍、四肢瘫痪、胰腺炎、免疫性损伤、视神经萎缩等[22]。在治疗过程中,若患儿出现其他系统的损害,可采取对症处理,贫血患儿可补充叶酸、维生素B12,脑积水患儿可采取脑脊液引流手术治疗[10]。MMA的预后主要取决于诊断、分型及治疗的时间,患儿起病时间越早,病情越重,死亡率也越高[23]。
因此,新生儿串联质谱筛查、尿有机酸和基因检测对于MMA的早期诊断起到了至关重要的作用。本研究通过对18例mut型MMA患儿的临床资料进行总结分析,对mut型MMA的诊断和治疗提供了一定的帮助,希望未来能出现更多更准确的早期诊断方法,使患儿尽早得到有效治疗,有利于改善患儿预后,从而降低患儿的致残率、死亡率。
