雷贝拉唑钠肠溶片的溶出度研究及方法学验证*
2022-03-11孟长虹汪玉馨陆益红
赵 悦,张 营,杜 益,严 菲,孟长虹,汪玉馨**,陆益红**
1 徐州医科大学 药物分析教研室,徐州 221004;2 江苏省食品药品监督检验研究院,南京 210019;3 中国人民解放军联勤保障部队第988 医院,郑州 450000;4 浙江省中医院 药剂科,杭州 310006
质子泵抑制剂(proton pump inhibitors,PPI)是治疗消化性溃疡的首选药物,也是目前抑制胃酸分泌作用最强的一类药物。雷贝拉唑钠肠溶片(rabeprazole sodium enteric-coated tablets)是第二代PPI,可通过抑制部分细胞分泌表面的H+/K+-ATP 酶抑制胃酸分泌、而不影响胆碱能受体或组胺H2受体[1],在临床上用于消化系统溃疡及返流性食管炎、卓艾氏综合征等的治疗[2]。雷贝拉唑钠是雷贝拉唑的钠盐,由日本卫材公司成功研发,于1997 年首先在日本上市,随后分别在欧洲、美国、中国上市。雷贝拉唑钠为弱碱性药物,在酸性环境中不稳定[3],可被胃酸迅速降解失活,因此,为增加药物稳定性,提高生物利用度,目前其口服制剂多开发为肠溶片或肠溶胶囊。
药物进入人体后,活性成分的释放速度与程度是影响药物吸收快慢的主要因素[4]。溶出度试验是一种模拟固体口服制剂在胃肠道中崩解和溶出的体外试验方法,是评价口服固体制剂质量的重要指标。溶出曲线是根据药物在不同时间的溶出量绘制出的曲线[5],故通过测定不同溶出介质(如pH 1.2、pH 6.0、pH 6.8、水)体外溶出曲线的相似性,评价国内仿制药与原研药的差异。目前有文献报道[6],采用规定的高效液相色谱法测定雷贝拉唑钠肠溶片的溶出曲线,但雷贝拉唑钠肠溶片在偏中性溶出介质中不稳定,在pH 6.0 溶出介质中45 min 时几乎完全降解,在pH 6.8 溶出介质中45 min 时有部分降解,其测定的是雷贝拉唑钠在溶出过程中降解后的溶出量,故该法不能客观评价该产品的溶出情况。
紫外分光光度法-等吸收点-单波长法和等吸收点-双波长法在排除降解产物、制剂辅料等的干扰,准确进行含量测定方面具有一定的优势。王艺红等[7]利用等吸收点-双波长分光光度法测定盐酸普鲁卡因注射液的含量,排除了制剂中对氨基苯甲酸(PABA)对普鲁卡因含量测定的干扰,解决了亚硝酸钠法不能排除制剂制备储存过程中的分解产物PABA 对测定结果影响的问题。孙长霞等[8]将等吸收点-双波长的定量理论应用于紫外-可见分光光度法中,可快速检测口蘑中微量铜离子,也排除了干扰物吸收或散射的干扰。
本研究建立了紫外分光光度法-等吸收点-双波长吸收差值法测定雷贝拉唑钠肠溶片的溶出度,并进行了方法学验证,对国内A 厂、B 厂的雷贝拉唑钠肠溶片仿制药与卫材(中国)生产的参比制剂进行对比研究,为客观评价雷贝拉唑钠肠溶片国内仿制药与原研药溶出曲线的相似性提供依据。
1 仪器与药品、试剂
1.1 仪器
SNTR-8400AT 型自动溶出仪、LC-20AB 泵高效液相色谱仪(岛津仪器有限公司);8510 型超声波清洗器(美国Branson 公司);XP6 型/XS205DU 型电子天平、UV5Nano 型紫外分光光度计(梅特勒-托利多仪器有限公司);DTC-41 型真空脱气泵(ULVAC KIKO 公司)。
1.2 药品与试剂
参比制剂[卫材(中国),规格:10 mg、20 mg,批号:1602031、1604013];仿制制剂(A 厂,规格:10 mg,批号:170202、151113、160105;B 厂,规格:20 mg,批 号:20160705、20160303、20160302);雷贝拉唑钠对照品(纯度99%,批号:100658-210404,中国食品药品检定研究院);氯化钠、盐酸、氢氧化钠、无水磷酸氢二钠、一水合柠檬酸、磷酸二氢钾,均为分析纯;乙腈为色谱纯;水为纯化水。
2 方法与结果
2.1 溶液的配制
根据《普通口服固体制剂溶出曲线测定及比较指导原则》及相关文献[9,10],选择pH 1.2、pH 6.0、pH 6.8 介质和水4 种溶出介质测定雷贝拉唑钠肠溶片的溶出度。
2.1.1 溶出介质的配制①pH 1.2 溶出介质:取氯化钠2.0 g,加水适量使溶解,再加盐酸7 mL,加水稀释至1000 mL;②pH 6.0 溶出介质:取磷酸氢二钠17.93 g,柠檬酸7.74 g,加水适量使溶解,加水稀释至1000 mL;③pH 6.8 溶出介质:取磷酸二氢钾1.7 g,无水磷酸氢二钠1.775 g,加水适量使溶解,加水稀释至1000 mL;④水。
稀释溶液:分别取0.5 mol·L-1NaOH-pH 1.2(1∶3)、0.5 mol·L-1NaOH-pH 6.0(1 ∶3)、0.5 mol·L-1NaOH-pH 6.8(1∶3)与0.5 mol·L-1NaOH-水(1∶3)作为稀释溶液1、2、3、4。
2.1.2 对照品储备液的配制精密称定雷贝拉唑钠对照品4 份,每份20 mg,置10 mL 量瓶中,分别加稀释溶液1、2、3、4 振荡溶解并定容至刻度,作为雷贝拉唑钠对照品储备液1、2、3、4。
2.2 溶出量测定方法的确定
2.2.1 HPLC 法测定精密称定4 份雷贝拉唑钠对照品,每份10 mg,分别加入900 mL pH 1.2、pH 6.0、pH 6.8 介质和水4 种溶出介质中,水浴(37±0.5)℃,分别于5、10、15、20、25、30、35、45、60、90、120、180 min取出3mL 溶液,立即加入0.5mol·L-1NaOH 溶液1mL,混匀(因雷贝拉唑钠在酸性条件下,会迅速降解,故需加入一定量的碱性溶液使其稳定)。按照高效液相色谱法测定[11]。
色谱柱:X-Bridge C18(4.6 mm×150 mm,5 μm);流动相:0.015 mol·L-1磷酸二氢钠(pH 值7.0)-乙腈(35∶65);检测波长:290 nm;柱温:30 ℃;流速:1.0 mL·min-1;进样量:10 μL。见图1。
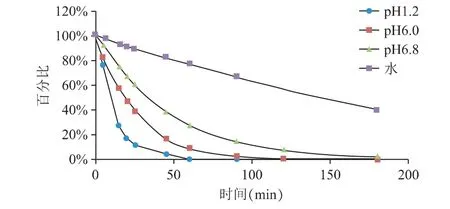
图1 雷贝拉唑钠肠溶片在pH 1.2、pH 6.0、pH 6.8 介质和水4 种溶出介质中随时间降解曲线
由图1 可知,雷贝拉唑钠在pH 1.2、pH 6.0、pH 6.8 介质和水4 种溶出介质中均不稳定,在放置过程中发生降解,25 min 时pH 1.2、pH 6.0、pH 6.8 介质和水4 种溶出介质中降解率分别为88%、61%、39%和11%,降解结果见表1。

表1 雷贝拉唑钠随时间降解速率(%)
雷贝拉唑钠肠溶片在模拟小肠吸收的介质(pH 6.0、pH 6.8)中溶出过程不稳定,当药物在溶出介质中释放后,会显现出主成分降解的趋势[12],故该法不能测定雷贝拉唑钠溶出过程中降解部分的量,无法获得完整的溶出曲线。
为解决此难题,本研究基于降解产物与主成分具有相似的母核结构,在紫外吸收中存在等吸收点的情况,采用紫外分光光度法-等吸收点-双波长吸收差值法测定雷贝拉唑钠肠溶片在溶出介质中的累积溶出量。
2.2.2 紫外-可见分光光度法及检测波长的选择按“2.2.1”项下方法,分别于5、10、15、20、25、30、35、45、60、90、120、180min 取3mL 溶液,立即加入0.5mol·L-1NaOH 溶液1 mL,混匀。按紫外-可见分光光度法在200~600 nm 进行全波长扫描,结果见图2。
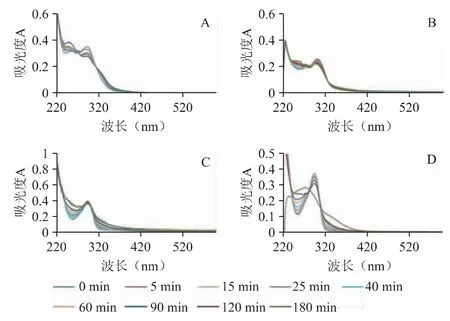
图2 雷贝拉唑钠在pH 1.2(A)、pH 6.0(B)、pH 6.8(C)、水(D)4 种溶出介质中全波长扫描图
从图2 紫外扫描发现,不论雷贝拉唑钠在4 种溶出介质中如何降解,雷贝拉唑钠在pH 1.2 溶出介质中有277 nm 和330 nm 两个等吸收点;pH 6.0 溶出介质中有280 nm 和332 nm 两个等吸收点;pH 6.8 溶出介质中等吸收点为290 nm;水中有285 nm和303 nm 两个等吸收点。因330 nm、332 nm、303 nm属于边缘吸收波长,会对测定结果产生影响,故选择277 nm 为雷贝拉唑钠在pH 1.2 溶出介质中的测定波长;280 nm 为雷贝拉唑钠在pH 6.0 溶出介质中的测定波长;290 nm 为雷贝拉唑钠在pH 6.8 溶出介质中的测定波长;285 nm 为雷贝拉唑钠在水中的测定波长。
2.2.3 双波长吸收差值测定法的确定雷贝拉唑钠本身在特定波长处有较好吸收,但肠溶制剂中不仅存在主成分,其他成分也可能对有效成分的紫外分光光度分析产生干扰。如其辅料中含有苯环和双键结构,则会干扰有效成分的分析[13],影响紫外分光光度法测定雷贝拉唑钠肠溶片溶出度的准确性。因此,若单一使用等吸收点作为其溶出度的检测波长,会存在一定误差,故需选择远端主成分无吸收的波长(400 nm),同时测定等吸收点和400 nm 波长处的吸光度,用吸光度差值ΔA 计算雷贝拉唑钠肠溶片的溶出量,以抵消干扰组分在两个波长处产生的吸光度,消除溶出测定过程中的辅料干扰,同时也不会对雷贝拉唑钠的测定产生影响。
取A 厂、B 厂及参比制剂各一批样品4 片,分别置于100 mL 量瓶中,分别加入稀释溶液1、2、3、4 适量,超声溶解并稀释至刻度,最终均稀释为10 μg·mL-1的样品溶液(n=3)。同时取对照品储备液1、2、3、4,稀释成10 μg·mL-1的溶液,作为对照品溶液。分别采用等吸收点单波长法与等吸收点双波长法,以单波长下吸光度A 或双波长下吸光度之差ΔA,计算雷贝拉唑钠肠溶片的含量,结果以标示量的百分比表示,见表2。

表2 单波长法与双波长法含量测定结果比较(为标示量%)(n=3)
采用《中国药典》2015 年版二部本品“含量测定” 项下的HPLC 法,A 厂、B 厂及参比制剂含量测定平均值分别为标示量的100.8%、99.2% 及101.5%。由表2 可知,按单波长测定法在4 种溶出介质中含量测定结果平均值分别为标示量的107.0%、107.2%及110.4%,按双波长测定法含量测定平均值分别为标示量的100.3%、100.1%及101.6%。
采用SPSS 25.0 软件对数据进行统计学分析,组间比较,采用独立样本t 检验,单波长测定法与HPLC法比较,差异有统计学意义(t=5.947,P=0.004),双波长测定法与HPLC 法比较,差异无统计学意义(t=0.201,P=0.850),故选择双波长吸收差值法。
2.3 溶出曲线研究方法
取雷贝拉唑钠肠溶片12 片,按溶出度与释放度测定法(2015 版《中国药典》四部通则0931 第二法)测定[11],在pH 6.8 溶出介质条件下,溶出介质体积为900 mL,温度为(37±0.5)℃,转速为50 r·min-1、100 r·min-1。分别于5、10、15、20、25、30、35、45、60、90 min取样,用0.45 μm 微孔滤膜滤过,取续滤液3 mL,加入0.5 mol·L-1NaOH 溶液1 mL,混匀,按等吸收点-双波长吸收差值法计算累积溶出量,结果见图3。
由图3 可知,在转速100 r·min-1条件下,雷贝拉唑钠肠溶片溶出较快,15 min 时溶出度已达94.8%,而在转速50 r·min-1条件下15 min 时为22.9%,可见100 r·min-1条件过于剧烈,故本实验选取转速50 r·min-1。
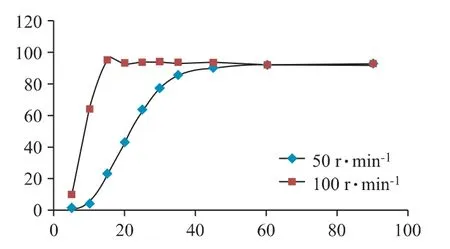
图3 雷贝拉唑钠肠溶片不同转速下溶出曲线图(n=12)
2.4 溶出度测定方法学验证
2.4.1 线性关系考察精密称取雷贝拉唑钠对照品4 份,每份10 mg,置10 mL 量瓶中。加入0.5 mol·L-1NaOH 溶液溶解并定容至刻度,精密量取1 mL,分别加入3 mL 对应的溶出介质,作为雷贝拉唑钠对照品储备液,再用稀释溶液1、2、3、4 依次稀释成不同浓度的对照品溶液。以浓度C 为横坐标,吸光度差值ΔA 为纵坐标,吸光度差值ΔA 与药物浓度C 计算所得回归方程及相关系数r,见表3。由表3可知,雷贝拉唑钠在0.20~27.30 μg·mL-1浓度范围内线性关系良好。
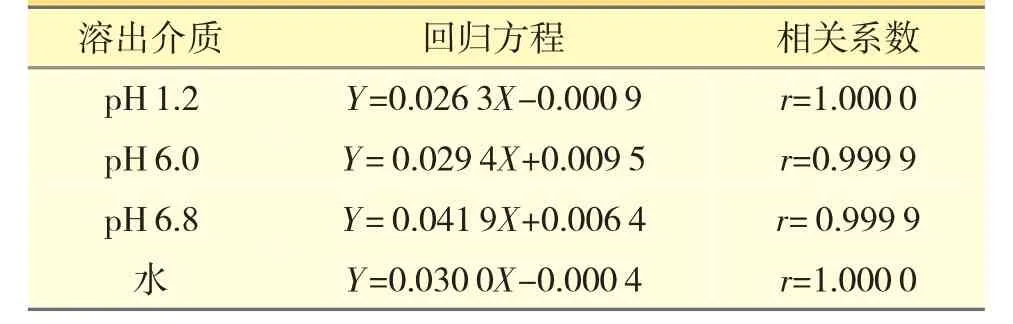
表3 4 种溶出介质标准曲线回归方程
2.4.2 精密度试验精密称取雷贝拉唑钠对照品4份,每份10 mg,置100 mL 量瓶中,分别加入适量稀释溶液1、2、3、4,超声溶解并定容至刻度,再精密量取1 mL 置10 mL 量瓶中,分别用稀释溶液1、2、3、4稀释至刻度。按照紫外-可见分光光度法在pH 1.2、pH 6.0、pH 6.8 介质和水4 种溶出介质中,于277、280、290、285 和400 nm 波长处测定对应的吸光度,连续测定6 次,计算等吸收点和400 nm 波长处的吸光度差值ΔA。RSD 值分别为1.32%、0.27%、0.70%、0.25%,表明该法的精密度良好。
2.4.3 稳定性试验精密称取雷贝拉唑钠对照品4份,每份10 mg,分别加入pH 1.2、pH 6.0、pH 6.8 介质和水4 种溶出介质900 mL(37 ℃),搅拌并溶解均匀,37 ℃水浴中放置,于0、5、15、25、40、60、90、120、180 min 取样3 mL,分别加入0.5 mol·L-1NaOH 溶液1 mL,于277、280、290、285 和400 nm 波长处测定吸光度。雷贝拉唑钠在4 种溶出介质中各时间点吸光度差值ΔA 的RSD 分别为0.85%、0.71%、0.91%、0.95%,表明雷贝拉唑钠在4 种溶出介质中180 min 内吸光度稳定,无显著变化。
2.4.4 重复性试验取雷贝拉唑钠肠溶片细粉,精密称取适量(约含雷贝拉唑钠10 mg)置100 mL 量瓶中。加适量稀释溶液1,超声溶解并定容至刻度,摇匀滤过,精密量取续滤液1 mL 置10 mL 量瓶中,加稀释溶液1 定容至刻度。依此方法平行配制6 份样品溶液,按照紫外分光光度法,测定277 nm 等吸收点及400 nm 两处波长的吸光度。其余3 种溶出条件同pH 1.2 介质溶出条件下方法,分别计算280、290、285 nm 等吸收点与400 nm 波长处吸光度差值ΔA。雷贝拉唑钠在4 种溶出介质中的RSD 分别为0.87%、0.86%、0.66%、1.04%(n=6),表明该法的重复性良好。
2.4.5 回收率试验精密称取3 份已知含量的雷贝拉唑钠肠溶片细粉(约含雷贝拉唑钠5 mg),分别置50 mL 量瓶中。分别加入对应的对照品储备液2.0、2.5、3.0 mL,加入对应的稀释溶液,超声溶解并定容至刻度,摇匀滤过,精密量取续滤液1 mL 置10 mL量瓶中。以上步骤平行操作3 次,按照紫外分光光度法,于277、280、290、285 和400 nm 波长处测定吸光度。分别计算277、280、290、285 nm 等吸收点与400 nm 波长处吸收度差值ΔA,计算平均回收率和RSD(n=3),计算结果见表4。由表4 可知,在4 种溶出介质中,雷贝拉唑钠肠溶片的回收率的RSD 均<2%,表明该法的准确度良好。
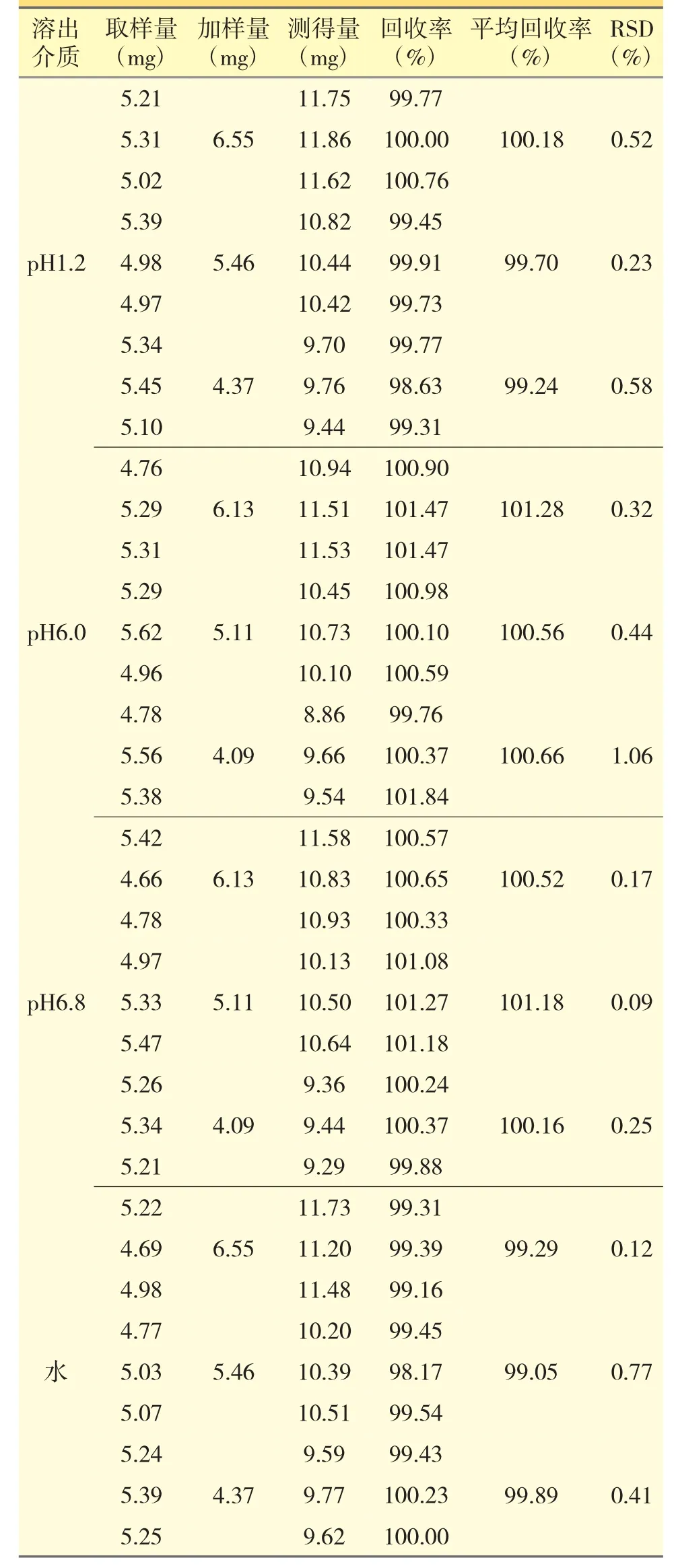
表4 雷贝拉唑钠肠溶片在不同溶出介质中回收率实验结果(n=9)
2.5 溶出度测定
取A 厂、B 厂、卫材(中国)雷贝拉唑钠肠溶片各12 片,按照溶出度与释放度测定法(2015 版《中国药典》四部通则0931 第二法)测定[11],以pH 1.2、pH 6.0、pH 6.8 介质和水4 种溶液作为溶出介质,溶出体积为900 mL,温度为(37±0.5)℃,转速为50 r·min-1。依法操作,分别于5、10、15、20、25、30、35、45、60、90、120min 取溶液10mL,取样后补充相同温度和体积的溶出介质。取出溶液用0.45 μm 微孔滤膜滤过,取续滤液3mL,加入0.5mol·L-1NaOH 溶液1mL,混匀。按照等吸收点-双波长吸收差值法计算各时间点的累积溶出百分率,以溶出时间为横坐标,累积溶出率为纵坐标,绘制相应的溶出曲线。见图4。
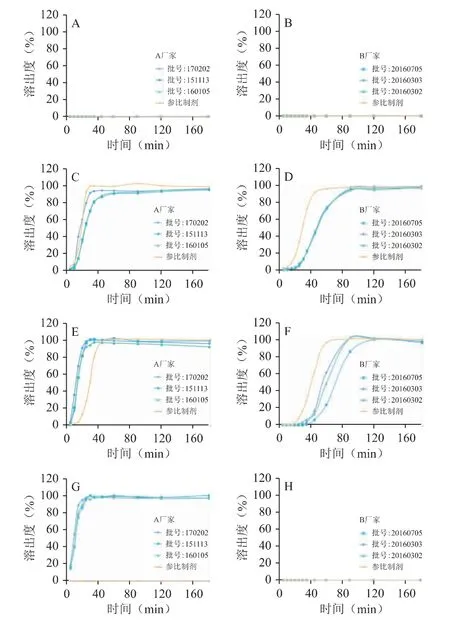
图4 参比制剂与A 厂(规格:10mg)、B 厂(规格:20mg)各3批制剂在pH 1.2、pH 6.0、pH 6.8 介质和水4 种溶出介质中的溶出曲线
3 讨论
雷贝拉唑钠在酸性及中性介质中不稳定,故将其制成肠溶制剂。但由于雷贝拉唑钠肠溶片在偏中性溶出介质中(pH 6.0,pH 6.8)也会发生降解,HPLC法只能测定雷贝拉唑钠肠溶片主成分的量,不能完整、客观表征雷贝拉唑钠肠溶片的溶出和释放过程,且药物辅料在测定过程中也会产生干扰。故本研究建立了紫外分光光度法-等吸收点-双波长吸收差值法,测定雷贝拉唑钠肠溶片在4 种溶出介质中的累积溶出量。从抗酸能力(pH 1.2)和溶出曲线两方面可见,A 厂、B 厂肠溶包衣均具有较好的抗酸能力,可以确保样品到达肠道后溶出。采用日本PMDA(pharmaceuticals and medical devices agency)推荐的pH 6.0 和pH 6.8 溶出介质进行溶出曲线的绘制,可进行相似性评价。
建立的方法解决了雷贝拉唑钠肠溶片在酸性、尤其是中性介质中累积溶出量测定的问题,消除了辅料干扰紫外吸收的情况。
通过方法学验证,该法具有良好的准确度、精密度、重复性,可分析雷贝拉唑钠肠溶片在酸性及中性介质中的溶出度,指导仿制药的处方、工艺开发和质量控制,也为其他药物在生理溶出介质中溶出度不稳定、辅料成分干扰大的问题提供了解决思路。
由于口服肠溶制剂在人体胃肠道中除有释放过程外,还存在药物吸收过程,用体外溶出的方法预测体内吸收过程具有一定的局限性,故还应关注药物在体内的吸收情况。体外溶出曲线研究结合体内生物等效性系统评价雷贝拉唑钠肠溶片,目前国内研究较少,本研究拟进行体内外相关试验,建立参比制剂与仿制药体外溶出曲线与体内生物等效性之间的相关性模型,以探索体外溶出条件与药物在体内释放行为的相关性。
