亮菌甲素注射剂杂质谱研究
2022-03-11薛敏华康丽洁石蓓佳陆益红
尹 菁,薛敏华,康丽洁,石蓓佳,陆益红*
1 江苏省食品药品监督检验研究院,南京 210019;2 阜阳市第五人民医院,安徽阜阳 236063
亮菌甲素又称假蜜环菌甲素,是一种香豆素类化合物,具有利胆解痉、松弛胆道末端括约肌作用,临床作为利胆药[1]。其剂型有片剂、注射剂。亮菌甲素、注射用亮菌甲素及亮菌甲素注射液的现行标准均收载于《国家药品标准》[2-4],其中亮菌甲素原料药采用TLC 法控制有关物质,注射用亮菌甲素及亮菌甲素注射液现行标准中未设置有关物质检查项;亦有注射用亮菌甲素执行企业注册标准[国家食品药品监督管理局标准(试行)YBH08512006],有关物质检查未拟定有效的系统适用性指标,难以确保杂质峰的有效分离与检出[5]。目前尚未见关于亮菌甲素杂质谱的研究报道。本研究采用液质联用技术,运用强制降解试验,建立了亮菌甲素注射剂杂质谱,对10个杂质结构进行定性分析并推测杂质来源。此外,采用ADMET Predictor 软件对亮菌甲素及其杂质进行毒性预测,为亮菌甲素的质量控制提供参考。
1 仪器与药品、试剂
1.1 仪器
Acquity UPLC I CLASS,Xevo G2-XS Q-TOF液相质谱联用仪(MassLynx V4.1/UNIFI 分析软件,美国Waters 公司);高效液相色谱仪LC-20AD,SPD-M20A 检测器(日本Shimadzu 公司);Mettler Toledo XS205 型电子天平(瑞士Mettler Toledo 公司,感量:0.01mg);SevenMulti 型pH 计(瑞士Mettler Toledo 公司);7.2.0001 型DMET Predictor 药物预测软件(美国Simulation Plus 有限公司)。
1.2 药品与试剂
亮菌甲素原料(a 厂,批号20150927);注射用亮菌甲素(b 厂,批号:17041222、18011132、18031113),亮菌甲素注射液(c 厂,批号:1702102、1702112、1702122);乙酸铵、乙酸、氢氧化钠等为分析纯;盐酸、30%过氧化氢为市售品;甲醇为色谱纯;水为超纯水。
2 方 法
2.1 色谱条件
色谱柱:Kromasil 100-5-C18(250 mm×4.6 mm,5 μm);流动相:0.01 mol·L-1乙酸铵溶液(用乙酸调pH 至4.0)为流动相A,甲醇为流动相B,梯度洗脱:0~20 min B 由10%升至40%,20~40 min B 由40%升至60%,40~50 min B 保持60%,50~51 min B 降至10%,51~65 min B 保持10%;柱温:35 ℃;检测波长:340 nm;流速:1 mL·min-1;进样量:10 μL。
2.2 质谱条件
离子化模式:电喷雾离子源(ESI-);毛细管电压:3.0 kV/2.5 kV;源温度:120 ℃;雾化气流速:1000 L·h-1;雾化气温度:500 ℃;采集模式:MSE(全信息串联质谱)。
2.3 溶液的制备
2.3.1 有关物质供试品溶液避光操作。①亮菌甲素原料:称取10 mg 置于20 mL 量瓶,用甲醇溶解,加水稀释至刻度,摇匀;②注射用亮菌甲素:取本品,用水溶解并稀释制成每1 mL 中约含亮菌甲素0.5 mg的溶液;③亮菌甲素注射液:取本品进样分析。
2.3.2 亮菌甲素原料强制降解试验①酸破坏:称取亮菌甲素10 mg 置于20 mL 量瓶,加1 moL·L-1盐酸2 mL,室温暗处放置1 h,用1 moL·L-1氢氧化钠调至中性,加甲醇溶解,用水稀释并定容,摇匀。②碱破坏:称取亮菌甲素10 mg 置于20 mL 量瓶,加0.1 moL·L-1氢氧化钠2 mL,室温暗处放置30 min,用0.1 moL·L-1盐酸调至中性,加甲醇溶解,用水稀释并定容,摇匀。③氧化破坏:称取亮菌甲素10 mg置于20 mL 量瓶,加30%过氧化氢溶液2 mL,室温暗处放置1 h,加甲醇溶解,用水稀释并定容,摇匀。④高温破坏:称取亮菌甲素10 mg 置于20 mL 量瓶,于60 ℃烘箱放置10 天,取出至室温,加甲醇溶解,用水稀释并定容,摇匀。⑤光照破坏:称取亮菌甲素10 mg 置于20 mL 量瓶,加甲醇溶解,用水稀释并定容,摇匀,置强光下照射2 h,照度(4500±500)Lx。
2.4 杂质毒性预测
使用Chemdraw 软件画出亮菌甲素及各杂质的结构式并保存为“.mol”或“.sdf”格式,导入ADMET Predictor 软件,对亮菌甲素及各杂质的毒性作出预测。
3 结果
3.1 亮菌甲素杂质谱的建立
对b、c 厂生产的亮菌甲素注射剂进行有关物质检查,典型色谱图见图1A、B。取“2.3.2”项下各溶液进样分析,典型色谱图见图1C。强制降解试验结果表明,亮菌甲素在碱性条件下极不稳定,在光照条件也不稳定。在选定的色谱条件下,各杂质色谱分离良好。将原料、制剂中存在的杂质以及强制降解试验产生的的主要杂质汇总,并绘制成亮菌甲素的杂质谱,见图2。10 个主要杂质峰按保留时间顺序编号为杂质1~10,其中杂质1~5 为碱降解杂质,杂质6、7 为光降解杂质,杂质8 为原料引入的,杂质9、10 为亮菌甲素注射液特有的杂质。

图1 亮菌甲素注射剂有关物质及原料降解试验色谱图
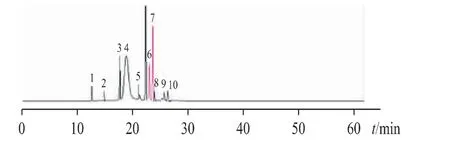
图2 合成的亮菌甲素杂质谱
3.2 亮菌甲素注射剂中主要杂质LC-MS 分析
根据质谱信息,采用Masslynx V4.1/NNIFI 软件中的化合物结构解析工具预测杂质分子式,作元素分析及化合物鉴定;参考相关文献[6]并依据各杂质来源及亮菌甲素裂解规律,推定各杂质结构[7]。该品杂质谱中10 个杂质质谱信息与推测结构见表1,图3。

图3 亮菌甲素的杂质结构式
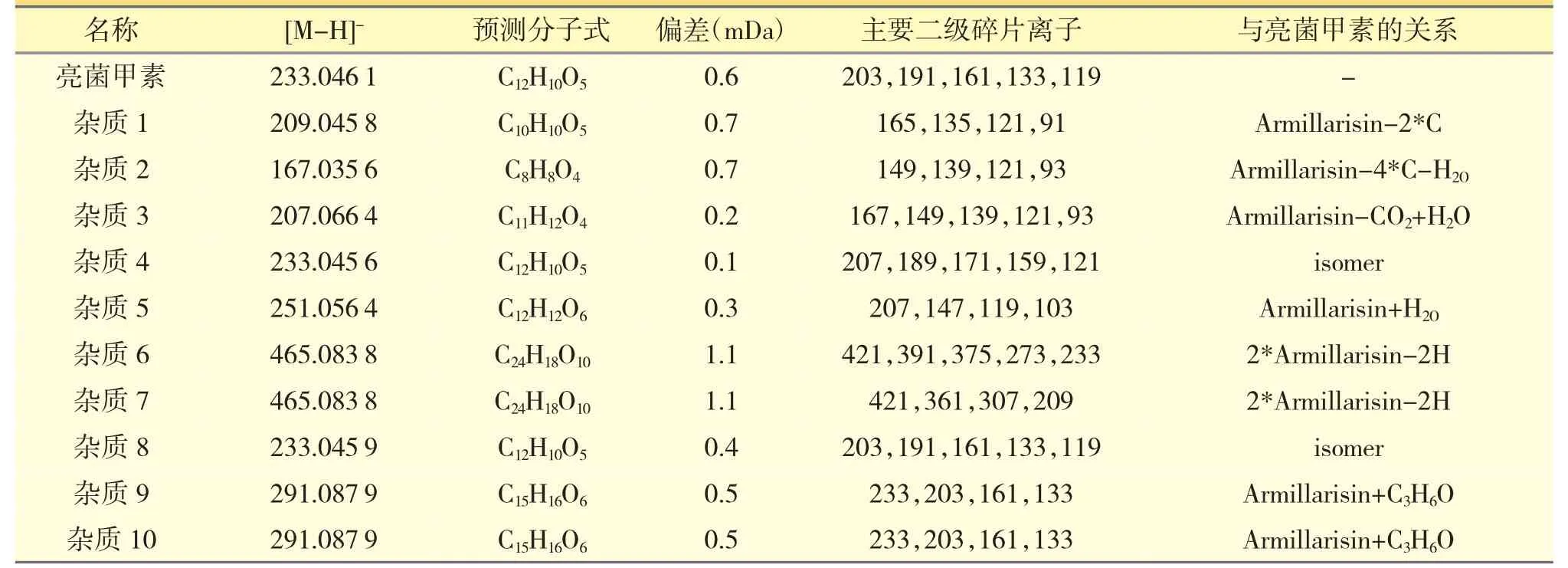
表1 亮菌甲素杂质质谱信息与推测的结构
3.2.1 亮菌甲素裂解途径推测亮菌甲素的[M-H]-分子离子为m/z 233,主要的二级碎片离子m/z 203,m/z 191,m/z 161,m/z 133,m/z 119。先通过丢失苯环上的羟甲基形成碎片离子m/z 203 [M-HCH2O]-,或通过丢失内酯环上的乙酰基形成碎片离子m/z 191[M-H-C2H2O]-,若同时通过丢失苯环上的羟甲基和内酯环上的乙酰基,则形成碎片离子m/z 161[M-H-CH2O-C2H2O]-,最后通过丢失内酯环中一分子CO 形成碎片离子m/z 133[M-H-CH2O-C2H2OCO]-,或通过丢失内酯环中一分子CO2形成碎片离子m/z 119[M-H-CH2O-C2H2O-CO2]-。根据其典型的碎片离子推测亮菌甲素质谱裂解途径,见图4。

图4 亮菌甲素质谱裂解途径推测
3.2.2 杂质裂解途径推测①杂质1 的结构分析:杂质1 的[M-H]-准分子离子峰为m/z 209,软件推测分子式为C10H10O5。其二级碎片离子与亮菌甲素不一致,说明其基本母核与亮菌甲素不同。杂质1的质量比亮菌甲素少24,结合杂质1 的来源,推测杂质1 是由亮菌甲素碱破坏导致内酯环断开进而丢失2C。碎片离子m/z 165 是由母离子m/z 209 苯环上取代基侧链丢失一分子CO2形成的;碎片离子中有m/z 135,说明苯环上有羟甲基;该离子进一步丢失甲基形成碎片离子m/z 121,最后形成稳定的碎片离子m/z 91。因此,杂质1 的结构及可能的质谱裂解途径推测见图5。
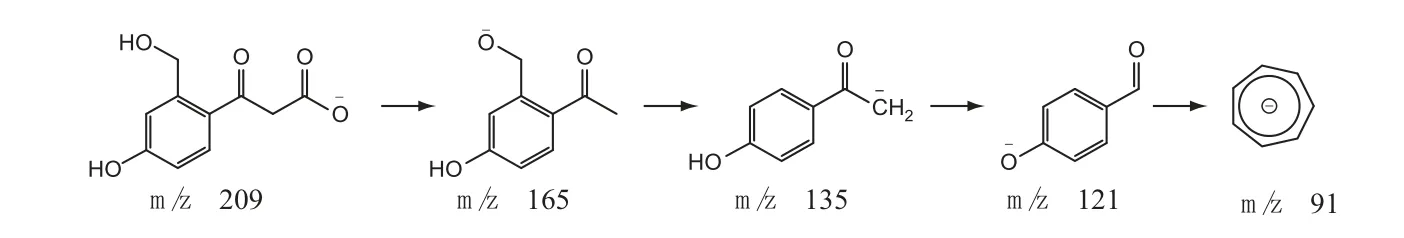
图5 杂质1 的结构及可能的质谱裂解途径
②杂质2 的结构分析:杂质2 的[M-H]-准分子离子峰为m/z 167,二级碎片离子与亮菌甲素不同,说明其基本母核与亮菌甲素不同。杂质2 的质量比亮菌甲素少66,结合杂质2 的来源及分子式预测为C8H8O4,推测杂质2 是由亮菌甲素内酯环断开后丢失4C 和一个中性分子H2O 形成的。m/z 149 是由[C8H8O4]-发生C-O键断裂丢失一个中性分子H2O 形成的,再通过丢失甲酰基形成[C7H5O2]-碎片离子m/z 121;m/z 139 是由[C8H8O4]-丢失醛基形成的,进而发生C-O 键断裂形成[C7H5O2]-碎片离子m/z 121;最后[C7H5O2]-丢失一个中性CO 分子得到碎片离子m/z 93。杂质2 结构及推测的质谱裂解途径见图6。

图6 杂质2 的结构及可能的质谱裂解途径
③杂质3 的结构分析:杂质3 的[M-H]-准分子离子峰为m/z 207,二级碎片离子中m/z 149,m/z 139,m/z 121,m/z 93 与杂质2 相同,其基本母核可能与杂质2 一致,即亮菌甲素中内酯环断开,这与杂质3 是由亮菌甲素经碱破坏或高温破坏得到的一致。杂质3 的质量比杂质2 多40,碎片离子中有m/z 165 但是没有m/z 167,推测杂质3 苯环侧链有乙酰基;结合杂质3 的来源及Masslynx V4.1/NNIFI软件分析结果,推测杂质3 的结构及可能的质谱裂解途径见图7。
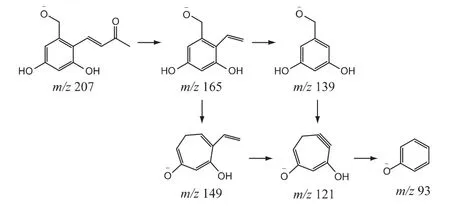
图7 杂质3 的结构及可能的质谱裂解途径
④杂质4 的结构分析:杂质4 的[M-H]-准分子离子峰为m/z 233,分子式预测结果为C12H10O5;杂质4 的一级质谱信息与亮菌甲素基本一致,推测为亮菌甲素的同分异构体,但其二级质谱信息与亮菌甲素不相似,推测其空间结构可能与亮菌甲素不同。由于亮菌甲素母核有内酯环,在碱性条件下易发生水解、内酯环断开,加酸中和后内酯环重新环合;若羧基与苯环上邻位羟基脱水环合则形成亮菌甲素的内酯环,推测杂质4 的内酯环是由羧基与邻位羟甲基上的羟基脱水环合形成的。因此,推测杂质4 的结构及可能的质谱裂解途径见图8。
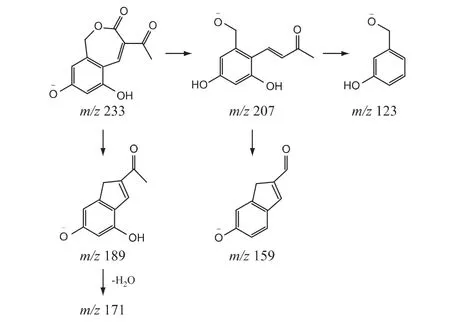
图8 杂质4 的结构及可能的质谱裂解途径
⑤杂质5 的结构分析:杂质5 的[M-H]-准分子离子峰为m/z 251,分子式预测结果为C12H12O6;其二级碎片离子与亮菌甲素不一致,说明其基本母核与亮菌甲素不同。杂质5 的质量比亮菌甲素多18,推测其结构中引入H2O;杂质5 二级碎片离子中m/z 207 是由母离子丢失羧基形成的,进一步发生CO 键断裂丢失一个中性H2O 分子,同时丢失乙酰基后形成碎片离子m/z 147,该碎片离子再丢失一个中性CO 分子形成碎片离子m/z 119,最后失去氧原子形成碎片离子m/z 103。结合Masslynx V4.1/NNIFI 软件分析结果,推测杂质5 的结构及可能的质谱裂解途径见图9。
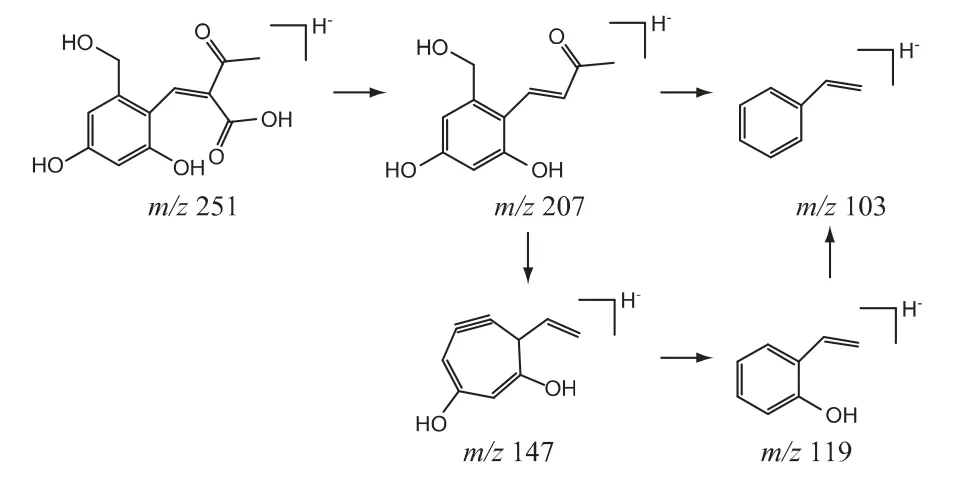
图9 杂质5 的结构及可能的质谱裂解途径
⑥杂质6、杂质7 的结构分析:杂质6、7 的[MH]-准分子离子峰m/z 均为465,软件推测分子式为C24H18O10,且杂质6、7 的一级质谱信息相似,推测杂质6、7 互为为同分异构体[8]。但杂质6 与杂质7 的二级质谱信息不一致,推断这两个杂质的空间结构不同;结合强制降解试验,表明杂质6、7 为光降解杂质,推测未知杂质6、7 为亮菌甲素二聚体;根据化合物极性关系及洗脱顺序,推断杂质6 极性较杂质7 大,故先被洗脱出峰。故杂质6、杂质7 结构及推测的质谱裂解过程见图10 和图11。

图10 杂质6 的结构及可能的质谱裂解途径

图11 杂质7 的结构及可能的质谱裂解途径
⑦杂质8 的结构分析:杂质8 的[M-H]-准分子离子峰为m/z 233,分子式预测结果C12H10O5,推测为亮菌甲素的同分异构体;杂质8 与亮菌甲素的二级质谱信息基本一致,可见杂质8 的空间结构与亮菌甲素相似。经杂质来源分析可知,杂质8 为原料引入的。推测杂质8 为亮菌甲素原料合成中3,5-二羟基苯甲醇与乙氧亚甲基乙酰乙酸乙酯缩合反应的的副产物,结合Masslynx V4.1/NNIFI 软件分析结果,杂质8 的结构及可能的质谱裂解途径见图12。

图12 杂质8 的结构及可能的质谱裂解途径
⑧杂质9、杂质10 的结构分析:杂质9、10 的[M-H]-准分子离子峰m/z 均为291,分子式预测结果为C15H16O6;杂质9、10 二级质谱信息基本一致,故为同分异构体;杂质9、10 的质量比亮菌甲素多58,推测其分子中引入C3H6O。杂质9、10 为亮菌甲素注射液中特有的杂质,推测杂质9、10 为亮菌甲素注射液中辅料与亮菌甲素反应的产物。为验证上述推论,称取亮菌甲素5 mg,按处方比例,加1,2-丙二醇3 mL与水7 mL,混匀,105 ℃热处理1 h(模拟生产工艺),取出至室温。按有关物质检查的色谱条件进样分析,在相对保留时间(RRT)1.18 和1.21 处出现了两个杂质峰,证明杂质9、10 是亮菌甲素与辅料丙二醇在制剂过程中发生反应所得,为工艺杂质。杂质9、10 的结构及可能的质谱裂解途径见图13。
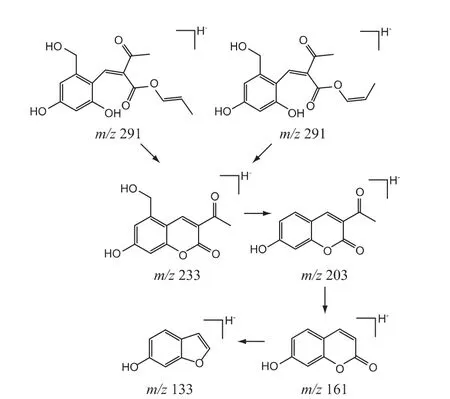
图13 杂质9、10 的结构及可能的质谱裂解途径
3.3 杂质来源分析
结合强制降解试验、杂质结构解析结果以及生产工艺分析,推测各杂质来源见表2。
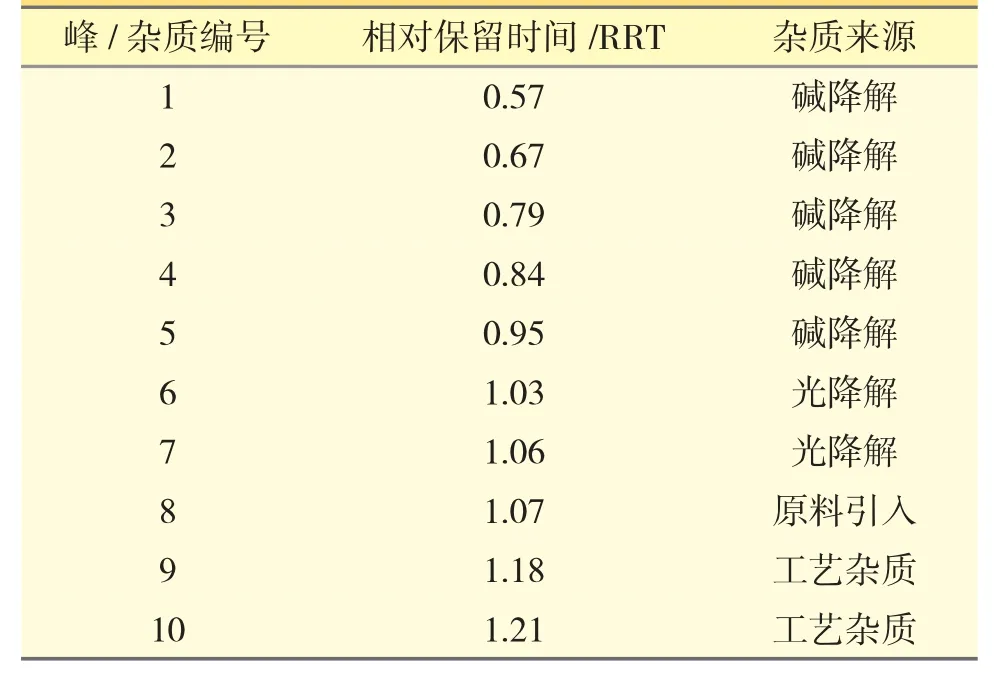
表2 亮菌甲素注射剂杂质及来源归属
亮菌甲素注射剂在制剂过程中,采用先在碱性条件下使亮菌甲素溶解,再用酸回调的工艺。杂质1~5 为亮菌甲素在碱性条件下内酯水解开环及进一步降解产生的一系列杂质。碱的强度以及在碱性条件下保持的时间长短会影响杂质1~5 产生的量。杂质6、7 为亮菌甲素的光降解杂质。杂质8 为原料引入的,推测为原料合成3,5-二羟基苯甲醇与乙氧亚甲基乙酰乙酸乙酯缩合反应的副产物。杂质9 和10为亮菌甲素注射液特有的杂质,从LC-MS 预测的分子式推测为占处方比例30%的辅料——丙二醇与亮菌甲素反应的产物。
3.4 亮菌甲素注射剂中主要杂质毒性预测
利用ADMET Predictor 软件对亮菌甲素及10个杂质的ADMET 性质做了全面评价[9],杂质毒理预测结果见表3。其中亮菌甲素、杂质8 的预测毒性很低,风险很低;其他杂质可能具有一定的潜致敏性、染色体畸变、生殖毒和肝脏不良反应,但风险较低。

表3 ADMET Predictor 软件预测结果
4 讨论
强制降解试验表明,亮菌甲素光照敏感,在强碱性条件下极不稳定。两种剂型产品中均未检出光降解杂质,说明企业在生产、储存过程中的避光措施合理。由于亮菌甲素在水中几乎不溶,生产工艺中均是将该品溶解于碱性溶液中,再用酸调节pH,说明控制产品的pH 值是制备的关键点,建议产品在生产过程中应尽量缩短与碱接触的时间,严格控制pH。亮菌甲素注射液因辅料中使用大量的丙二醇,而产生了该剂型特有的杂质9、10,应改进处方工艺,降低丙二醇引入的杂质。
本研究采用液质联用技术,同时结合强制降解试验结果以及亮菌甲素注射剂的处方和生产工艺,推测了亮菌甲素注射剂杂质谱中10 个杂质的结构与来源,并用计算机软件预测了10 个杂质的毒性,为该品种的处方和生产工艺优化、质量控制及储存条件提供了参考依据。
