抗病毒核苷酸类似物磷酸化修饰研究进展
2022-03-10梁星星王佳许文涛
梁星星 王佳 许文涛
(1. 中国农业大学营养与健康系,北京 100091;2. 中国农业大学食品科学与营养工程学院,北京 100083)
核苷(酸)是构成生命遗传信息的基础物质,参与细胞内生物大分子DNA和RNA的合成,同时还以不同形态参与人体内大量的生化反应过程,如细胞的信号传递,能量代谢,以及各种酶的调节与表达等。核苷(酸)类似物是一类合成的、经化学修饰的,模仿核苷(酸)生理学性质的物质,它们可以参与到细胞内的代谢过程,嵌入到DNA及RNA中阻断其延伸复制,从而抑制细胞的生长和增殖[1]。核苷(酸)类似物已广泛应用于抗病毒治疗,如由人类免疫缺陷病毒(human immunodeficiency virus,HIV)、疱疹病毒(herpes virus,HPV)、乙型肝炎病毒(hepatitis B virus,HBV)、丙型肝炎病毒(hepatitis C virus,HCV)等引起的疾病[2-3],并且,瑞德西韦(remdesivir,一种腺嘌呤核苷类似物前药)对治疗最近爆发的SARS CoV-2感染表现出很好的治疗前景[4]。
核苷(酸)类似物通过与内源核苷(酸)相同的代谢机制而被激活,其首先通过细胞膜上特定的核苷(酸)载体(如一些转运蛋白载体、有机阴离子和阳离子交换蛋白等)进入细胞[5],随后在细胞内激酶的作用下逐步磷酸化,生成具有生物活性的核苷三磷酸类似物,这些活化后的核苷酸类似物可竞争性抑制病毒编码的DNA或RNA聚合酶的作用,如HIV逆转录酶、HCV编码的RNA聚合酶等;也可直接参与病毒内DNA/RNA的合成,嵌入其中并导致链延伸的终止,从而发挥抗病毒作用[3,6]。由于核苷(酸)类似物的第一步(或第二步)磷酸化是由病毒编码的激酶催化完成的,这也保证了这些药物只作用在被病毒感染的细胞中[7]。接下来的磷酸化步骤由宿主细胞内的激酶催化完成,由于核苷(酸)类似物的性质不同,其细胞内的磷酸化过程可能需要一种、两种、三种甚至更多酶来催化完成,不同核苷(酸)类似物与激酶的亲和力也各不相同,激酶在细胞内的含量和有效性也各不相同[8-9]。因此,激酶催化是整个反应的限速步骤,其催化过程经常不充分,导致最后生成的核苷三磷酸类似物在细胞内的含量偏低,降低了药物的生物活性。因此,开发高度磷酸化的核苷酸类似物是很有必要的,它们可以避开细胞内的限速步骤(即激酶催化),提高细胞内生成的核苷三磷酸类似物的浓度,从而提高抗病毒治疗效果。然而,未修饰的核苷酸类似物在生理条件下带负电荷从而显示出极性,使其不易穿过细胞膜;其次,核苷酸类似物在体内易受磷酸酶的降解而脱磷酸[10]。为改善和解决上述问题,研究人员开发了多种合成方法,如通过在核苷酸类似物上修饰亲脂性基团,以确保其不易被血液中的磷酸酶降解,同时增强被细胞摄取吸收的能力,最终实现高效的抗病毒作用[11-13]。根据磷酸化程度的不同以及核苷酸类似物的发展过程,可将核苷酸类似物分为3种:核苷单磷酸、双磷酸和三磷酸类似物。
目前,已通过美国食品药品监督管理局(FDA)或欧洲药品管理局(EMA)的核苷(酸)药物有替诺福韦酯(tenofovir dipivoxil)、司他夫定(stavudine)、伐昔洛韦(valacyclovir)、西多福韦(cidofovir)等,并且已被批准用于治疗人类疾病[14]。本文总结概述了核苷酸类似物在抗病毒药物领域的研究进展,以磷酸化修饰为主,重点介绍了核苷单磷酸、双磷酸和三磷酸类似物的发展,并分析了核苷酸类似物在临床药物应用中存在的局限和未来发展的趋势,以期为开发新型、高效的核苷酸药物提供有益参考。
1 核苷单磷酸类似物
核苷单磷酸类似物是在核苷类似物基础上修饰单磷酸基团得到的核苷酸类似物,它能避开细胞内的第一步磷酸化步骤,在一定程度上提高抗病毒效果。然而由于其生理电负性和脱磷酸化作用,极大地限制了该类物质的应用。因此,为保证药物能顺利进入细胞,并不易被血液中的磷酸酶降解,需要对磷酸基团上带的负电荷进行掩蔽,常见的修饰基团有bisPOM-、SATE-、CycloSal-和氨基磷酸酯等[15-16],通过这些方法制备的中性亲脂性核苷单磷酸三酯能通过被动运输进入细胞并在酶解或者化学水解作用下生成相应的具有生物活性的核苷单磷酸类似物。
早在1983年,Farquhar等[17]就报道了双-碳氧甲基修饰的磷酸酯衍生物在肝酯酶和血浆中具有很好的稳定性。随后,该团队又利用中性双-酰氧基甲基修饰核苷单磷酸,其能有效提高药物的亲脂性,结构如图1所示。该药物易于进入细胞,并能在羧酸酯酶作用下裂解产生羟甲基核苷单磷酸中间体,该中间体继续发生裂解,生成核苷单磷酸[18]。双-酰氧基甲基修饰的核苷单磷酸具有很好的稳定性,因此其在体内循环过程中不易被降解,提高了其到达细胞内的有效浓度。利用双-新戊酰氧基甲基[bis(pivaloyloxymethyl),bis(POM)]修饰的无环核苷单磷酸类似物具有广谱的抗病毒活性[19],对bis(POM)修饰的5-叠氮基甲基2′-脱氧尿苷(AmdU)的代谢合成进行研究发现,其可以高效地活化为核苷三磷酸形式从而合成到DNA中,且在细胞培养基中展示了较高的稳定性[20]。
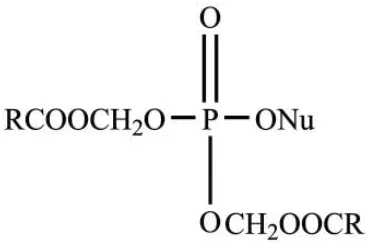
图1 双-酰氧基甲基核苷单磷酸Fig. 1 Bis-acyloxymethyl nucleoside monophosphate[18]
与双-酰氧基甲基修饰类似,S-酰基-2-硫乙基(S-acyl-2-thioethyl,SATE)基团修饰的核苷单磷酸类似物的水解也是依赖酯酶激活,接着发生自发裂解。根据酸碱理论,对于巯基乙醇取代的磷酸衍生物,巯基会进攻β-C进而使巯基乙醇从磷酸上水解下来,形成核苷酸。基于此,Peyrottes等[21]采用SATE基团作为核苷单磷酸的保护基团,其结构如图2所示(R表示烷基或者芳基,Nu表示核苷类似物),SATE基团在细胞外基质中有很好的稳定性,而在细胞内可以快速地降解,这有利于在细胞内选择性地释放核苷单磷酸药物,从而提高抗病毒效果。作为一个生物可裂解的磷酸保护基团,该双-SATE-核苷单磷酸在细胞内酯酶的介导下水解生成不稳定的磷酸三酯,接着发生自发裂解,最后得到具有生物活性的核苷单磷酸。体外实验表明,双-SATE-磷酸三酯衍生物的抗病毒活性均高于未修饰的核苷。SATE法可广泛用于核苷单磷酸类似物的修饰,如双-(叔丁基-SATE)-阿糖胞苷(bis(tBuSATE)-araC)[22],双-(叔丁基SATE)-7-芳基取代的7-脱氮杂腺苷[23]等。
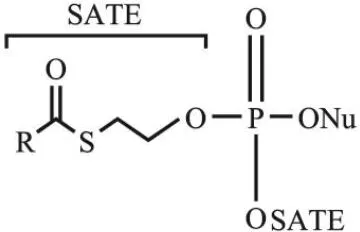
图2 Bis-(SATE)-核苷磷酸三酯的结构Fig. 2 Structure of bis-(SATE)-nucleoside phosphotriester[21]
除了依靠酶解产生活化的核苷酸药物外,还可以依赖化学裂解来激活核苷单磷酸药物。CycloSal-核苷酸是由水杨醇与核苷单磷酸反应形成的三酯化合物,由苯基酯键和苄基酯键组成环化双功能基团,以2′,3′-双脱氢-2′,3′-双脱氧胸腺嘧啶核苷单磷酸(d4TMP)为例,其结构如图3所示,CycloSal-核苷酸在细胞内的裂解完全依赖pH驱动的化学裂解。由于水杨醇的修饰掩蔽了磷酸基团上的单键氧,因此得到的CycloSal-核苷酸呈中性,且亲脂性大大提高。相比于2′,3′-双脱氢-2′,3′-双脱氧胸腺嘧啶核苷(d4T),CycloSal-d4TMP能够通过被动运输穿过细胞膜,并且成功地避开了第一步磷酸化,使得最终活化后的d4TTP的浓度显著提高[24]。利用CycloSal的亚氯酸盐与核苷反应可生成相应的CycloSal修饰的核苷单磷酸,基于此方法,Kamata等[25]合成了CycloSal修饰的4′-乙炔基-2′-脱氧腺苷(EdA)磷酸酯衍生物,其可有效地抑制HBV的复制,且对宿主细胞无毒。CycloSal-核苷酸的抗病毒活性不仅与核苷及取代基团的种类有关,其立体构象也显著影响着抗病毒活性。以甲基-取代的CycloSal-d4TMP为例,其抗病毒活性强烈依赖于磷酸基团及CycloSal基团中甲基的手性,不同立体构象的CycloSal-d4TMP的抗病毒活性可以相差7-20倍[26]。
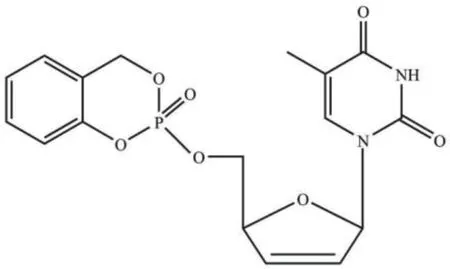
图3 CycloSal-d4TMP的结构Fig. 3 Structure of CycloSal-d4TMP[24]
此外,氨基磷酸酯也是一类常用的核苷单磷酸修饰基团,以P-N键代替P-O键,结合氨基酸侧链修饰,不仅能有效稳定磷酸基团,同时有助于增强药物的抗病毒活性[27]。例如,芳香基和氨基磷酸甲酸酯修饰的无环核苷单磷酸对DNA病毒有很好的抗性[28];通过氨基磷酸酯修饰合成的2′-β-甲基-2′-α-氟尿苷单磷酸(sofosbuvir,索非布韦)可用于治疗丙型肝炎病毒(HCV)引起的疾病[29];最近研究较多的瑞德西韦(remdesivir)是一种氨基磷酸酯修饰的单磷酸前药,研究表明,其可有效地抑制SARSCoV-2在人肺细胞培养物中的复制[30]。
核苷单磷酸类似物胞内传递的实现推动了核苷酸类似物在体内的应用,通过在磷酸基上修饰亲脂性基团,提高了核苷单磷酸类似物的稳定性和膜穿透性。双-酰氧基甲基修饰的核苷单磷酸药物中有两种已用于临床治疗,替诺福韦酯(tenofovir dipivoxil)和阿德福韦酯(adefovir dipivoxil),可分别用于治疗HIV和HBV感染。bis(SATE)修饰虽然可以提高核苷单磷酸药物的亲脂性,但是SATE基团裂解的过程中会产生副产物硫化乙烯,该物质对机体有毒性,因此bis(SATE)修饰的核苷单磷酸药物的使用剂量应尽可能降低,以减小对机体的伤害。CycloSal-法合成的核苷单磷酸类似物的脱磷酸化过程是pH依赖型的,无需酶的参与,且只需要一个激活步骤,由于CycloSal-基团的双功能性,合成过程中CycloSal-基团与核苷酸分子的比率为1∶1,从而没有副产物的产生[12],是修饰核苷酸类似物常用的一种方法。氨基酸侧链修饰得到的核苷单磷酸前药的抗病毒潜力与氨基酸种类有关,通过使用不同的氨基酸侧链,可以调整核苷单磷酸前药的生物活性。此外,CycloSal-法也可用于合成核苷双磷酸类似物[16,31],氨基磷酸酯修饰的核苷单磷酸前药大部分也实现了在临床上的应用[32-33],因此,近几年关于CycloSal法和氨基磷酸酯修饰的研究报道也较多。目前用于临床研究或治疗的核苷单磷酸药物除了阿德福韦酯和替诺福韦酯外,还有帕拉德福韦(pradefovir)、索菲布韦(sofosbuvir)、CMX-001(brincidofovir)、贝西福韦(besifovir)、ODE(HDP)-(S)-HPMPA、MCC-478和USC-087等[34]。
2 核苷双磷酸类似物
核苷单磷酸类似物虽然避开了细胞内的第一步磷酸化,但对于某些核苷酸类似物来说,第二步的磷酸化仍是限速步骤,所以对于核苷双磷酸药物的研究是很有必要的。例如,第一个通过的抗HIV药物AZT,在AZT磷酸化为核苷三磷酸AZT-TP的过程中,由于第二步磷酸化(即AZT-DP的形成)仍是限速步骤,导致了AZT-MP的累积,因此产生了一些不良副作用[35]。然而,由于磷酸酐键的不稳定性,且磷酸基团只有在带负电荷的状态下才可以阻止亲核攻击而不裂解,因此核苷双磷酸类似物的稳定性较差,易被血清内非特异性磷酸酶水解而发生脱磷酸;另外,带负电荷的磷酸基团具有极性,这就导致核苷双磷酸类似物不易穿过细胞膜[36]。早期有许多关于核苷双磷酸类似物的研究,如利用双甘油酯[37-38]、肉豆蔻酸[39-40]和CycloSal[41]来修饰β-磷酸上的单键氧,这些方法在一定程度上提高了核苷双磷酸类似物的跨膜能力和稳定性,但其裂解的最终产物大部分都是核苷单磷酸,且伴随着一些副产物的产生,使细胞内有效的核苷双磷酸产物的浓度降低。
为保证磷酸酐键在最初的裂解反应中不受影响,采用酶解法取代化学裂解法来产生核苷双磷酸(NDPs)[41]。Weinschenk等[36]采用DiPPro法合成核苷双磷酸类似物,在β-磷酸上修饰两个一样的酰氧苄基酯,α-磷酸不修饰任何基团,因此核苷二磷酸类似物整体仍带负电荷,从而阻止磷酸酐键的裂解,保持焦磷酸酐的稳定,且不影响细胞吸收。其结构和水解途径如图4所示(R为烷基或者烯基),其中,第一步在细胞内酯酶的催化下,修饰基团的酯基发生裂解产生负电荷,接着其内部继续发生裂解,从而生成单酰氧基苄基修饰的核苷双磷酸,最后,再通过同样的机制生成最终产物NDPs。由于第一步酶催化的裂解不涉及磷酸酐键,因此可以成功地得到NDPs。
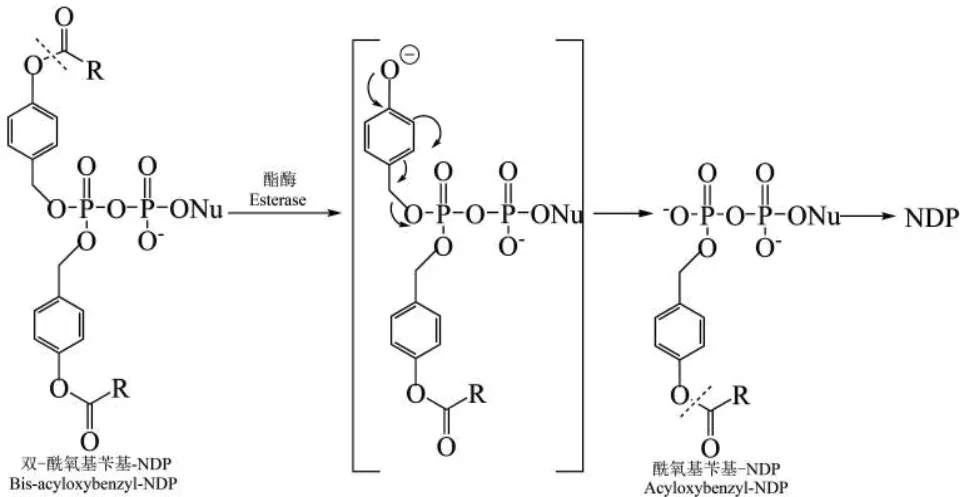
图4 DiPPro-药物的结构及水解路径Fig. 4 DiPPro-drug’s structure and hydrolysis path[36]
由于细胞内酯酶含量高,因此核苷双磷酸类似物的裂解活化主要发生在细胞内部。此法可用于合成一系列的核苷双磷酸药物,通过使用不同的取代基来调节核苷双磷酸类似物的稳定性和亲脂性[36-42]。但是,裂解产物中除了目标核苷双磷酸外,也检测到了少量的核苷单磷酸。因此,Weinschenk等[43]发展了第二代DiPPro-药物,利用两个不同的酰氧苄基酯来修饰β-磷酸,其中一个修饰基团是较短的烷烃链取代的酰氧苄基酯,酶解速度较快;另一个是较长的烷烃链取代的酰氧苄基酯或者苯基取代的酰氧苄基酯,以提高分子的亲脂性,此DiPPro-药物可以快速地裂解掉第一个修饰基团,从而使副反应产物NMPs减少,提高了药物的选择性。该方法有许多应用,如利用DiPPro法合成的T-1105核糖核苷双磷酸类似物抑制RNA病毒合成的效果要优于T-1105,因此提高了其抗病毒效果[44];除了用于抗病毒外,DiPPro法还可用于合成腺苷二磷酸核糖(ADPR)的前体物质,作为瞬时受体电位褪黑素2(TRPM2)的激活剂,DiPPro合成的ADPR衍生物可提高其激活效果,有望成为研究TRPM2的新方法[45]。
与核苷单磷酸药物不同的是,发展核苷双磷酸药物的关键在于保持磷酸酐键的稳定性。利用DiPPro-法合成核苷双磷酸类似物,仅对β-磷酸进行修饰,α-磷酸带的负电荷可稳定磷酸酐键,基于酯酶裂解发生脱磷酸化,由于细胞内的酯酶含量高且该药物在生理pH下可以保持很高的稳定性,因此可以保证NDPs只有在细胞内被释放。通过选择合适的取代基R,可以调节药物的稳定性和亲脂性,合成具有亲脂性的、带部分电荷的核苷双磷酸类似物,从而实现核苷双磷酸药物的传递。核苷双磷酸类似物适用于前两步磷酸化为限速步骤的核苷类似物,对于缺乏核苷双磷酸激酶活性的靶细胞来说,核苷双磷酸药物的抗病毒效果就会大大降低,因此只有传递核苷三磷酸类似物才可以解决此问题。
3 核苷三磷酸类似物
核苷单磷酸和双磷酸类似物的开发逐步实现了磷酸化形式核苷的递送,成功地避开了一步或两步磷酸化限速步骤,但释放的核苷酸仍需要通过细胞激酶进一步磷酸化成为核苷三磷酸才可发挥作用[46]。因此,直接开发核苷三磷酸药物具有重大优势和意义,它既能提高到达细胞内的活性药物浓度,也能减少细胞磷酸化过程中副产物的生成,从而有效提高治疗效果。然而,由于三磷酸基团带有4个负电荷,不易穿过细胞膜;且三磷酸基含有两个不稳定的磷酸酐键,其化学稳定性差,易被磷酸酶水解;同时,化学法合成核苷三磷酸药物也比较困难[47]。因此,核苷三磷酸类似物的发展极具挑战。
1995年,Bonnaffé等[48]首次提出制备酰基修饰的核苷三磷酸,通过酰基化增加其亲脂性,从而更易跨越细胞膜。以抗HIV的胸腺嘧啶核苷类似物d4T和AZT为研究对象,利用酰基化的焦磷酸与核苷磷酰吗啉反应制得了酰基化的NTPs。此外,Kreimeyer等[49]利用肉豆蔻酸与氯甲酸乙酯反应生成的酸酐与ADP或GDP反应,制备了嘌呤核苷三磷酸类似物。然而,通过以上两种方法制得的核苷三磷酸类似物产量极低,且易被水解。
随后,研究人员尝试仅对NTPs上的γ-磷酸进行酯化修饰,保留α-,β-磷酸的负电荷,在提高三磷酸基团的稳定性的同时,增加了NTPs的亲脂性,有利于其穿过细胞膜,使用不同的酰基酯可以调节NTPs的亲脂性、膜穿透性和稳定性[46,50]。Warnecke等[31]提出通过酯化反应合成酰氧苄基修饰的NTPs(TriPPPro法),利用不同的取代基团来调节核苷三磷酸类似物的亲脂性和化学稳定性,当该核苷三磷酸类似物进入细胞后,在酯酶作用下形成核苷三磷酸。该研究团队以d4T为研究对象,首先利用CycloSal技术合成了d4TDP,随后,此d4TDP与一系列含有两个相同的酰氧苄基修饰的亚磷酰胺单体反应,最后制得酰氧苄基修饰的d4TTP药物,其结构如图5所示(R为烷基、烯基、氧烷基和氨基烷基)[46]。
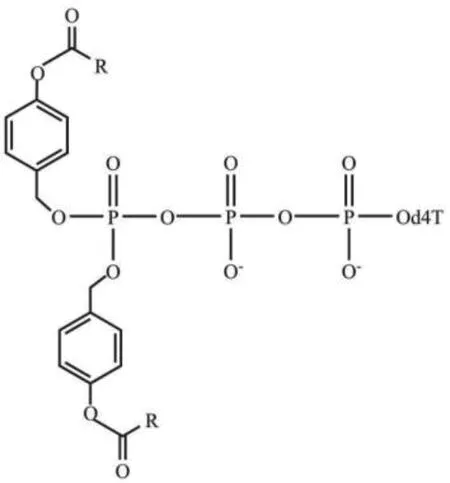
图5 酰氧苄基修饰的d4TTPFig. 5 Acyloxybenzyl modified d4TTP[46]
但是,该方法中核苷三磷酸类似物的最终产量仍然受到NDPs合成产率的限制。鉴于NMPs的制备相对NDPs较容易,该团队又研究了基于NMP和P,P-双-(4-壬酰氧苄基)修饰的焦磷酸反应生成TriPPPro-核苷酸类似物[9]。首先,采用CycloSal-法合成核苷单磷酸类似物[31],接着与P,P-双-(壬酰氧苄基)修饰的焦磷酸反应从而制得TriPPPro-核苷三磷酸类似物。利用此方法,Weising等[51]合成了抗HIV药物阿巴卡韦(abacavir,ABC)的亲脂三磷酸前药,活化后的ABCTP可作为dATP的竞争者参与DNA合成。
由于含碳酸酯的TriPPPro-核苷类似物的化学稳定性要高于含酰氧基的TriPPPro-核苷类似物,因此,Meier等[52]分别合成了酰氧苄基(acyloxybenzyl,AB,含酯基)和碳酸酯苄基(alkoxycarbonyloxybenzyl,ACB,含碳酸酯)修饰的核苷三磷酸类似物。通过研究γ-AB-ACB-d4TTPs和γ-ACB-ACB-d4TTPs在PBS溶液中的水解情况发现,γ-ACB-d4TTP的半衰期显著高于γ-AB-d4TTP,表明含碳酸酯修饰基团的药物具有更高的化学稳定性。此外,在γ-磷酸上修饰亲脂性的、生物可降解的ACB基团,增加核苷三磷酸的亲脂性,便于其穿过细胞膜。与γ-(AB,ACB)-d4TTPs不同,用两个不同长度的酰氧苄基修饰的d4TTPs在磷酸盐缓冲液和CEM细胞提取物中的水解速率并无差别[53];除了在γ-磷酸上修饰可降解的酰氧苄基外,还可修饰非裂解的烷基(alkyl),合成的γ-(AB,alkyl)-d4TTP在酯酶的作用下生成γ-alkyl-d4TTP,其可选择性地作为HIV逆转录酶的底物合成到DNA链中终止病毒复制,这种选择性使γ-(AB,alkyl)-核苷三磷酸特异性地作用于感染病毒的细胞中[54]。
虽然TriPPPro前药的磷酸基团仍带负电荷,但通过在γ-磷酸上修饰亲脂的、生物可降解的基团,以提供足够的亲脂性从而跨越细胞膜,并生成高含量的核苷三磷酸。通过在γ-磷酸上进行非对称的修饰,不仅可以同时提高TriPPPro前药的稳定性和亲脂性,而且可以实现修饰基团的逐步裂解,得到目标中间体。TriPPPro法合成的核苷三磷酸类似物可以完全避开细胞内磷酸化限速步骤,极大地提高了核苷酸药物的抗病毒效果,并且可以将无活性的核苷转化为有效的核苷酸类似物,在抗病毒和抗肿瘤应用中均有很好的应用前景,近几年的研究也主要集中在非对称修饰的TriPPPro前药[53-55],未来还需要进一步研究更加稳定、亲脂的核苷三磷酸前药,增强非天然的NTPs的在体内的传递效果,从而提高抗病毒核苷酸药物的治疗效果。
4 总结与展望
核苷酸类似物有很好的应用前景,本文概述了近几十年来抗病毒核苷酸类似物前药的研究进展,分别介绍了三类核苷酸类似物及其合成修饰方法,其主要通过对核苷酸进行磷酸化修饰各种亲脂性基团,合成可穿膜的具有活性的核苷酸类似物,在实现药物穿膜进入细胞的同时有效地缓解了血液中酶的脱磷酸作用,从而提高了进入细胞内核苷酸药物的浓度,最终生成具有活性的核苷三磷酸,从而发挥抗病毒作用。与核苷类似物不同,磷酸化修饰的核苷酸类似物可以避开细胞内的磷酸化限速步骤,有效地弥补了核苷类似物在应用中的局限性,提高了核苷酸药物的抗病毒效果,在一定程度上可以减少抗病毒药物的使用剂量,从而降低了机体对药物的敏感性。总的来说,核苷酸类似物具有高效的抗病毒应用潜力。
核苷酸药物的开发可以结合不同修饰基团的特点,如氨基磷酸酯基的稳定性和苄基的亲脂性,利用不同修饰基团对核苷酸进行磷酸化修饰,以提高核苷酸类似物的治疗效果。目前对于核苷酸类似物的开发研究比较多,但对于核苷酸类似物的细胞吸收机制以及吸收数量的研究还较少,修饰基团的种类对于抗病毒活性的影响也需要进一步的研究,通过不断优化修饰基团以达到更好的治疗效果。目前大多数的核苷酸类似物的裂解主要依靠酶解,核苷酸类似物在细胞内和血液中稳定性的差别大小决定了核苷酸类似物是否可以成功应用于体内,或许需要开发新的激活机制以提高核苷酸药物在血液中的稳定性和细胞内的裂解活性。另外,为了降低核苷酸药物对机体的毒副作用,核苷酸药物的使用剂量与抗病毒活性的关系还需要不断探索。
由于长期使用抗病毒药物会引起病毒的耐药性,同时为了降低机体对药物的敏感性,未来不仅需要进一步开发新型的、具有更高药效的抗病毒核苷酸类似物,更重要的是聚焦药物的低毒性、高水溶性和高特异性,开发具有低细胞毒性的药物递送系统,降低抗病毒药物的使用剂量,以减小机体对药物的敏感性。根据体内病毒水平选择不同强效的核苷酸药物,在病毒产生耐药性前换用另一种核苷酸药物,或者通过联合使用核苷酸药物,以降低病毒的耐药性,当然,规范用药,选择适当的核苷酸药物也是避免病毒产生耐药性重要措施。目前,超过100种抗病毒药物正在进行临床试验,但是大多数只针对一种病毒感染,新型的核苷酸药物还应具有针对多种病毒类型的广谱性。最后,要实现核苷酸类似物的广泛应用还需要进一步研究,如毒性试验,药物代谢动力学研究,临床试验以及如何实现大规模的生产等。
