高分辨质谱库和特征组分诊断比值法快速鉴别食品中非法添加淫羊藿
2022-03-07吴婉琴江丰干国平范小龙刘国姣王彬高芳朱松松张啟恒宋哲
吴婉琴,江丰,干国平,范小龙,刘国姣,王彬,高芳,朱松松,张啟恒,宋哲
(1.湖北省食品质量安全监督检验研究院,湖北 武汉 430075)(2.湖北省食品质量安全检测工程技术研究中心,湖北 武汉 430075)(3.湖北中医药大学药学院,湖北 武汉 430065)(4.湖北省中药炮制工程技术研究中心,湖北 武汉 430065)
淫羊藿(Epimedium brevicornuMaxim.)又名短角淫羊藿、仙灵脾等,为小檗科植物淫羊藿属淫羊藿的干燥叶,属应用历史悠久的常用补益中药,其具有益精气、补肾阳、强心力、坚筋骨等功效,可用于治疗风湿痹痛、筋骨萎软、阳痿遗精[1,2]。现代药理学研究表明,淫羊藿还具有促进缺血心肌血管新生、保护生殖系统、抗肿瘤、治疗类风湿关节炎、阿尔茨海默病等功能[3-9]。依据卫生部关于进一步规范保健食品原料管理的通知(卫法监发[2002]51号)[10]和《食品安全法》第三十八条[11],淫羊藿在药品及保健食品中允许添加,但在食品中禁止添加。一些食品生产企业受利益的驱动,只顾中药材功效而忽视其毒性和副作用,在所谓的“食疗”、“养生”等食品中违法添加淫羊藿,如宣称具有补益壮阳的配制酒、代用茶及饮料。在打击向普通食品违法添加中药材的行为过程中,监管部门执法人员只能通过查看产品配料表和到企业现场巡查等最传统的方式来判断食品中是否违法添加中药材,但往往会因生产企业不如实标注配料表或生产现场对非法添加的中药材原料进行隐藏等情况,形成监管盲区。因此,食品中非法添加中药材的鉴别工作在技术手段上面临巨大的挑战。
目前淫羊藿中药材的鉴别手段,主要有性状鉴别[12,13]、显微鉴别[14]、薄层鉴别[15,16]、中药材特征图谱[17,18]或指纹图谱鉴定[19,20]、DNA 条形码分子鉴定[21-23]等方法,但以上方法均是对中药材的真伪鉴别,而对食品中违法添加中药材的识别和鉴别,从本质上可追溯为鉴定其违法添加中药材的化学成分,而由于食品原料的多样性、食品基质和加工工艺的复杂性、加入的中药材品种未知性等原因,导致所添加的中药材在食品中可能与中药材原料中的特征图谱或指纹图谱发生了变化,因而以上几种鉴别方法不适用于食品中违法添加中药材的鉴别。
诊断比值(Diagnostic ratio,简称DR)是指样品中某些特定组分之间的比值,它能够表征不同样品各自的化学组成,用于判别两个样品来源是否一致[24]。其具有独特性和差异性,基本不受外界因素影响或受外界因素影响较小。目前主要运用于环境中污染物溯源鉴别[25-28]、海上溢油的鉴定[24,29,30]、医学疾病诊断[31-34]等领域,在食品领域用于中药材鉴别暂未见报道。
为了建立一种简单易行且可靠的鉴别食品中非法添加淫羊藿中药材的方法,本实验采用高效液相色谱-四极杆-飞行时间质谱仪构建淫羊藿特征组分高分辨质谱库进行初步筛查,再选取淫羊藿的6种特征成分(淫羊藿苷、朝藿定A、朝藿定B、朝藿定C、宝藿苷I、宝藿苷II)为考察对象,选取常见易非法添加中药材的食品配制酒、代用茶和饮料为研究对象,于高效液相色谱探索出基于特征组分的诊断比值法确证典型食品中违法添加淫羊藿中药材的鉴定技术,构建了食品中非法添加淫羊藿确证技术体系,为食品中淫羊藿非法添加的科学监管提供有效的技术支撑。
1 材料与方法
1.1 材料与仪器
淫羊藿中药材均购自电商平台,经干国平教授鉴定为小檗科植物淫羊藿Epimedium brevicornuMaxim.的干燥叶,采用粉碎机进行粉碎处理后过二号筛得淫羊藿粗粉,置于密封袋中,10批次淫羊藿中药材编号为 Y1-Y10,待检测;配制酒样品、代用茶样品、饮料样品,电商平台;淫羊藿对照药材,中国食品药品检定研究院;淫羊藿苷、朝藿定A、朝藿定B、朝藿定C、宝藿苷I、宝藿苷II(纯度≥98%),上海源叶生物技术有限公司;乙腈(色谱纯),德国Merck公司;甲醇(色谱纯),德国Merck公司;甲酸(质谱纯),美国 Fisher Scientific公司;超纯水(电阻率为 18.2 MΩ·cm,25 ℃),美国Millipore公司;其余试剂均为分析纯。
Ultimate 3000高效液相色谱仪,美国Thermo公司;AB Sciex TOF 5600+型串联四极杆质谱仪(配备Analyst 1.6工作站、Peak View定性筛查软件、Library View数据库软件),美国SCIEX公司;Waters e2695高效液相色谱仪、Waters 2998光电二极管阵列检测器,美国Waters公司;Allegra X-15R型离心机,美国Beckman公司;EDAA-2600T型超声波清洗器,上海安谱科学仪器有限公司;涡旋混合器,美国TALBOYS公司;ME2002E分析天平,梅特勒-托利多国际贸易上海有限公司;0.22 μm有机系滤膜、水系滤膜,天津津腾公司。
1.2 实验方法
1.2.1 标准溶液的配制
标准储备液:准确称取淫羊藿苷、朝藿定A、朝藿定B、朝藿定C、宝藿苷I、宝藿苷II标准品置于10 mL容量瓶中,用甲醇定容至刻度,摇匀,配制成淫羊藿苷、朝藿定A、朝藿定B、朝藿定C、宝藿苷I浓度为1.0 mg/mL,宝藿苷II浓度为0.5 mg/mL的标准储备液,置于4 ℃环境中冷藏避光保存。
标准工作溶液:分别取上述6种标准储备液适量,用甲醇稀释并定容,配制成浓度均为0.1 mg/mL的混合标准溶液,置于4 ℃环境中冷藏避光保存。
1.2.2 样品的制备
模拟淫羊藿酒的制备及前处理:称取淫羊藿中药材2 g(精确至0.01 g)置于棕色广口瓶中,准确加入20 mL 50%乙醇-水溶液,超声30 min,取出放至室温,浸提24 h以上。取上清液过0.22 μm有机系微孔滤膜,供高效液相色谱-四极杆-飞行时间质谱仪和高效液相色谱仪检测用。
模拟淫羊藿代用茶的制备及前处理:称取淫羊藿中药材2 g(精确至0.01 g)置于具塞离心管中,准确加入20 mL甲醇,超声30 min,取出放至室温,于4000 r/min离心5 min。取上清液过0.22 μm有机系微孔滤膜,供高效液相色谱-四极杆-飞行时间质谱仪和高效液相色谱仪检测用。
模拟淫羊藿饮料的制备及前处理:称取淫羊藿中药材2 g(精确至0.01 g)置于棕色广口瓶中,加入20 mL 50%乙醇-水溶液,超声30 min,取出放至室温,浸提24 h以上。于4000 r/min离心5 min,取上清液于圆底烧瓶,于 40 ℃水浴中减压浓缩至近干,准确加入20 mL超纯水,超声10 min,取上清液过0.22 μm水系微孔滤膜,供高效液相色谱-四极杆-飞行时间质谱仪和高效液相色谱仪检测用。
配制酒:取适量样品,于4000 r/min离心5 min,过0.22 μm有机系微孔滤膜,供高效液相色谱-四极杆-飞行时间质谱仪和高效液相色谱仪检测用。
代用茶:准确称取2 g(精确至0.01 g)样品置于具塞离心管中,准确加入20 mL甲醇,超声30 min,取出放至室温,于4000 r/min离心5 min。取上清液过0.22 μm 有机系微孔滤膜,供高效液相色谱-四极杆-飞行时间质谱仪和高效液相色谱仪检测用。
饮料:取适量样品,于4000 r/min离心5 min,过0.22 μm水系微孔滤膜,供高效液相色谱-四极杆-飞行时间质谱仪和高效液相色谱仪检测用。
1.2.3 仪器分析条件
1.2.3.1 HPLC-TOF-MS分析条件
(1)高效液相色谱条件
色谱柱:Thermo Accucore aQ 色谱柱(150×2.1 mm,2.6 μm);柱温:35 ℃;进样量:5 μL;流动相A为乙腈,流动相B为0.1%甲酸水溶液,梯度见表1。

表1 流动相梯度程序Table 1 Mobile phase gradient program
(2)TOF/MS工作条件
离子源:电喷雾离子源;正离子扫描模式:电喷雾电压:5500 V;离子源温度:550 ℃;气帘气:35 psi;雾化气:55 psi;辅助气:55 psi;去簇电压:60 V;碰撞能量:35 V;扫描方式采用全扫描一级质谱,质量采集范围100~1000 u;全扫描二级质谱,质量采集范围50~1000 u。质量数校正液为10 mmol/L甲酸钠溶液。
(3)TOF/MS筛查高分辨质谱库筛查条件
鉴别参数设置:化合物提取离子流响应强度>100或信噪比S:N>5,母离子精确分子量和二级碎片质量数偏差设为±(10×10-6),保留时间偏差±2.5%;数据库综合得分设置:质量数偏差、保留时间偏差、同位素比值、分子式匹配、数据库匹配占比均设为20%。
1.2.3.2 HPLC-PDA分析条件
色谱柱:Waters Symmetry Shield C18(250 mm×4.6 mm,5 μm);柱温35 ℃;检测波长270 nm;进样量:10 μL;流动相A为甲醇;流动相B为纯水,梯度见表2。乙酸铵溶液(0.1%乙酸)三种流动相体系分离效果。采用乙腈-纯水作为流动相时,淫羊藿苷、朝藿定A、朝藿定B、朝藿定C出峰较快导致分离度不理想;采用甲醇-20 mmol/L乙酸铵溶液(0.1%乙酸)时,6种化合物分离度较好,但是淫羊藿苷、朝藿定A、朝藿定B、朝藿定C有一定程度拖尾;采用甲醇-纯水时,6种目标化合物均有良好的分离度和峰形,如图1所示。故选择甲醇-纯水流动相体系进行分析。

表2 流动相梯度程序Table 2 Mobile phase gradient program
1.3 数据处理
采用AB Sciex Library View数据库软件构建目标化合物高分辨质谱库,采用AB Sciex Peak View定性筛查软件对样品进行筛查分析,采用SPSS 21.0统计软件对样品数据进行分析,采用SPSS 21.0统计软件和GraphPad Prism 5完成绘图。
2 结果与分析
2.1 仪器条件的优化
2.1.1 HPLC-TOF-MS
正离子扫描模式下,比较考察了乙腈-5 mmol/L乙酸铵和乙腈-0.1%甲酸两种流动相体系,部分化合物在乙腈-5 mmol/L乙酸铵流动相体系下峰形不佳出现拖尾现象,且峰响应较差,故选择乙腈-0.1%甲酸流动相体系进行分析。
为得到更全面的质谱数据信息,设定全扫描一级质谱,质量采集范围100~1000 u,全扫描二级质谱,质量采集范围50~1000 u。
2.1.2 HPLC-PDA
比较了甲醇-纯水、乙腈-纯水和甲醇-20 mmol/L
2.2 淫羊藿特征组分高分辨质谱数据库的建立
自建高分辨质谱库中淫羊藿包括6种特征成分一级精确质量数据库和二级碎片离子谱库,具体信息见表3。

表3 淫羊藿特征组分信息表Table 3 Information of characteristic components of epimedium
2.3 诊断比值的选取
结合上述配制酒、代用茶和饮料食品基质模拟过程,选用淫羊藿苷、朝藿定A、朝藿定B、朝藿定C、宝藿苷I、宝藿苷II 6种特征组分作为研究对象,通过各特征组分两两间峰面积的比值作为淫羊藿中药材特征组分的诊断比值,共计 15组诊断比值分别为DR1-DR15,如表4所示。

表4 淫羊藿中药材特征组分的诊断比值编号及描述Table 4 Diagnostic ratio number and description of characteristic components of epimedium
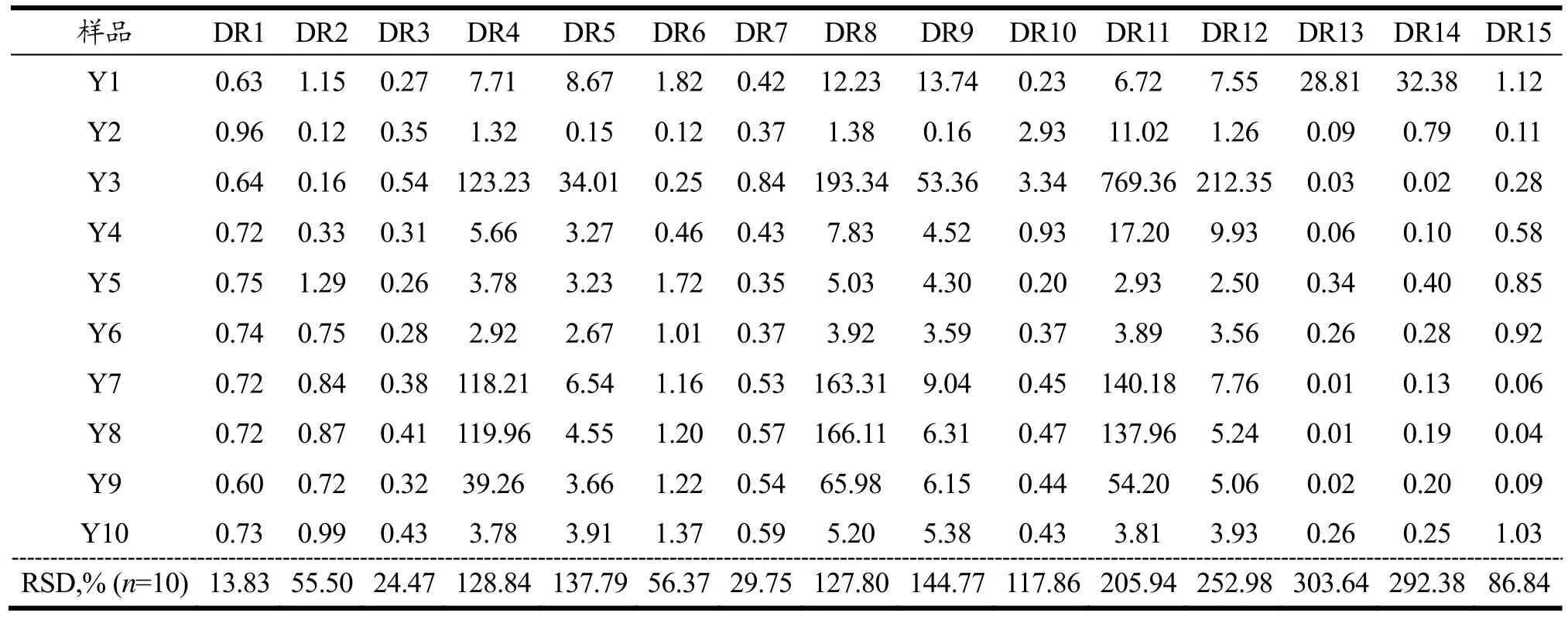
表6 10批次淫羊藿中药材模拟代用茶的特征组分诊断比值Table 6 Diagnostic ratio of characteristic components in 10 batches of substitutional tea of epimedium
对网购的10批次淫羊藿中药材样品按1.2.2过程处理后,各组分的诊断比值如表5~表7所示,在配制酒模拟过程中,朝藿定A与朝藿定B峰面积比值DR1数值集中在 0.63~0.94区间,各样品间的相对标准偏差(RSD)为12.49%;朝藿定A与淫羊藿苷峰面积比值DR3数值集中在0.27~0.64区间,各样品间的相对标准偏差(RSD)为29.30%;朝藿定B与淫羊藿苷峰面积比值DR7数值集中在0.37~0.87区间,各样品间的相对标准偏差(RSD)为35.00%。在代用茶模拟过程中,朝藿定A与朝藿定B峰面积比值DR1数值集中在 0.60~0.96区间,各样品间的相对标准偏差(RSD)为13.83%;朝藿定A与淫羊藿苷峰面积比值DR3数值集中在0.26~0.54区间,各样品间的相对标准偏差(RSD)为24.47%;朝藿定B与淫羊藿苷峰面积比值DR7数值集中在0.35~0.84区间,各样品间的相对标准偏差(RSD)为29.75%。在饮料模拟过程中,朝藿定A与朝藿定B峰面积比值DR1数值集中在0.61~0.86区间,各样品间的相对标准偏差(RSD)为10.53%;朝藿定A与淫羊藿苷峰面积比值DR3数值集中在 0.35~0.58区间,各样品间的相对标准偏差(RSD)为18.63%;朝藿定B与淫羊藿苷峰面积比值DR7数值集中在0.41~0.91区间,各样品间的相对标准偏差(RSD)为26.49%。通过各组诊断比值比较发现,在配制酒、代用茶和饮料三种模拟体系中,10批次样品的DR1,DR3,DR7数值波动相对较小,各样品间的相对标准偏差(RSD)均不高于35.00%,其它12组诊断比值波动区间较大,可能是由于受淫羊藿药材产地或采收季节的不同而造成了诊断比值的波动较大,故初步选择诊断比值DR1,DR3,DR7作为淫羊藿中药材鉴别的依据。

表5 10批次淫羊藿中药材模拟配制酒的特征组分诊断比值Table 5 Diagnostic ratio of characteristic components in 10 batches of prepared wine of epimedium

表7 10批次淫羊藿中药材模拟饮料的特征组分诊断比值Table 7 Diagnostic ratio of characteristic components of 10 batches of drink of epimedium
2.4 不同食品基质间诊断比值显著性差异分析
采用SPSS 21.0统计学软件对选定的模拟配制酒、代用茶及饮料食品的3组诊断比值DR1、DR3、DR7进行显著性差异分析,结果显示(见表8):3种食品基质间各组诊断比值均无显著性差异(p>0.05),表明所选的3组诊断比值具有一定的稳定性,不受样品基质、加工工艺和提取溶剂的干扰,可用于不同食品基质中淫羊藿中药材的鉴别。

表8 配制酒、茶叶和饮料食品的诊断比值方差分析Table 8 Analysis of variance of the diagnostic ratios of prepared wine, substitutional tea and drink
2.5 诊断比值非异常范围的确定
显著性差异分析显示3组诊断比值于不同基质间均无显著性差异,为更全面统一地对不同食品中淫羊藿进行鉴别,使方法具有一定的通用性,考虑将3种不同基质的各诊断比值进行合并,进行箱图绘制如图2,确定各比值非异常值范围,诊断比值DR1范围为0.60~0.96,诊断比值DR3范围为0.26~0.64,诊断比值DR7范围为0.35~0.91。
2.6 基于高分辨质谱库和特征诊断比值法对实际样品的检测
对购买的10批次淫羊藿中药材以及10批次配制酒、10批次代用茶和10批次饮料样品进行筛查分析,10批次淫羊藿中药材高分辨数据均与自建质谱库6种特征组分匹配一致,说明所建立的方法可有效筛查鉴别淫羊藿特征组分,1批次配制酒样品高分辨数据与自建质谱库6种特征组分匹配,推断该样品中可能含有淫羊藿。该配制酒样品靶向筛查结果如表9所示,配制酒样品中朝藿定A与标准品朝藿定A碎片离子镜像图如图3所示(图中上半部分为配制酒样品中朝藿定A的碎片离子图,下半部分为标准品朝藿定A的碎片离子图)。
为进一步确证样品中是否添加淫羊藿,采用特征组分峰面积诊断比值法对样品进行进一步分析。于高效液相色谱采用诊断比值法对该样品进行鉴别确证,检测出 6种特征组分,其中诊断比值 DR1(朝藿定A/朝藿定B)为0.73,诊断比值DR3(朝藿定A/淫羊藿苷)为0.28,诊断比值DR7(朝藿定B/淫羊藿苷)为0.38,3组诊断比值均在选定的诊断比值非异常值范围内,进一步表明该配制酒样品可能加入淫羊藿中药材。
目前淫羊藿中药材的鉴别手段,主要有性状鉴别、显微鉴别、薄层鉴别、中药材特征图谱或指纹图谱鉴定、DNA条形码分子鉴定等方法,研究对象多为单味药材饮片及复方制剂,对于食品中淫羊藿的鉴别鲜有报道,由于食品基质复杂且多样,成分组成繁多,对样品进行分析时相应的杂质干扰也多,单纯的性状鉴别、显微鉴别、薄层鉴别、DNA条形码分子鉴定对于复杂的食品基质中淫羊藿药材的鉴别已不适用,而中药材特征图谱或指纹图谱由于食品基质的干扰,在不同食品基质中会发生一定的变化,定性鉴别存在一定的偏差。对于食品中的淫羊藿的鉴别,要开发出一种不受不同基质影响且定性准确的方法,高效液相色谱-四极杆-飞行时间质谱仪通过测定母离子和碎片离子的精密质量数,定性准确度高,且分析速度快,用于多个化合物的初步筛查具有一定的优势,基于化合物定性准确的基础上,依据某种中药材自身特征组分之间不受其他因素影响具有的稳定比值即特征组分诊断比值法进一步定性,最终构建食品中淫羊藿药材的鉴别方法,所构建的方法较其它方法前处理简便,特征成分和诊断比值不受食品基质干扰和影响,可以准确地用于食品中淫羊藿的定性鉴别。
3 结论
本研究开发出一种基于高分辨质谱库和特征组分诊断比值法快速筛查鉴别食品中非法添加淫羊藿中药材的方法,选取6种特征组分构建高分辨质谱库进行初步筛查,最终确定稳定性较好的朝藿定A与朝藿定B峰面积比值(DR1)、朝藿定A与淫羊藿苷峰面积比值(DR3)、朝藿定B与淫羊藿苷峰面积比值(DR7)3组诊断比值作为鉴别依据,并考察了特征比值在配制酒、饮料、茶叶普通食品中的显著性差异,确定了3组诊断比值的非异常范围,构建了食品中淫羊藿中药材确证技术体系。该方法简便快捷、稳定、重复性好,可以有效地防止食品中淫羊藿中药材的非法使用,为打击食品中非法添加淫羊藿违法行为提供技术方法和依据。
