柱切换法液相色谱测定保健食品中的VA、VD、VE
2022-03-05张丽媛戚绿叶周明昊
张丽媛,戚绿叶,周明昊
(浙江省食品药品检验研究院,浙江 杭州 310052)
维生素是人体代谢中必不可少的有机化合物,是维持人体健康所必须的物质。这种物质在人体内不能合成或合成量太少,虽然需要量很少,但必须由食物供给[1]。保健食品是一种具有特定保健功能或者以补充维生素、矿物质为目的的食品,适宜于特定人群食用,具有调节机体功能的作用[2]。维生素类保健食品具有广泛的市场。VA又称视黄醇,能够促进骨骼生长,维持正常视力,缺乏可导致生长发育缓慢,干眼症等症状[3]。VD是一组具有抗佝偻病作用,结构类似的固醇类衍生物的总称,具有调节钙、磷代谢,促进骨骼发育的作用。最主要的是VD2(麦角钙化醇)和VD3(胆钙化醇)[4-6]。VA和VD都是脂溶性维生素,通常在食物中共存,主要存在于动物肝脏、蛋类、鱼肝油中。VA和VD在人体发育特别是对婴幼儿视力、生殖器官、骨骼、牙齿发育中的重要作用,促进了一系列维生素AD类保健食品的上市。VE又称生育酚,是重要的抗氧化剂,具有促生育、抗衰老,提高抵抗力的作用。生育酚主要有4种衍生物,按甲基位置分为α-、β-、γ-和δ-生育酚[7]。其中α-生育酚在自然界中分布最广泛,含量最丰富[8]。
目前,分离VE异构体的方法有薄层色谱法、气相色谱法和基于正相色谱的液相色谱法[9-12]。VD的测定方法有液相色谱法、液相色谱-质谱联用法、超临界流体色谱法、放射免疫与薄层色谱法等[13-15]。应用较广泛的为正相色谱净化、分离、反相色谱定量检测的液相色谱法。目前,对于VA、VD、VE类产品含量测定的研究,主要集中在婴幼儿配方乳粉以及配方饲料或中药方面[16-20],对于保健食品领域研究较少。保健食品中VA、VD、VE的测定主要依据GB 5009.82—2016《食品中维生素A、D、E的测定》。其中,VA、VE主要采用第一法,皂化萃取后用反相色谱定量检测;VD主要采用第四法,由于含量低,基质干扰严重,需采用正相色谱净化、分离、收集,反相色谱定量检测。同时测定VA、VD、VE需采用两个方法,两次前处理,两套液相色谱,对仪器、人员要求较高。整个实验过程繁琐复杂,多次转移复溶,且VA、VD、VE对光、热、氧气等较敏感,影响测定结果。
二维液相色谱基于其良好的分离效果,已广泛用于复杂基质样品中目标组分或全组分的分析[17]。二维液相色谱采用色谱柱串联技术,将样品净化、分离、收集、测定过程连续自动进行。样品经过一维色谱柱的初步分离,通过阀切换使其中一种或多种目标组分进入二维色谱柱中进一步分离,从而实现在复杂基质中排除干扰物质的在线净化分析过程[21-22]。随着二维液相色谱的发展普及,用于检测VA、VD、VE的文献也时有报道[11,16,19-25]。检测方法为,样品通过第一维色谱柱分离VA和VE,同时对VD进行净化,将初步净化后的VD在线引入第二维色谱柱进行分离测定,整个过程只需一次进样,便可完成样品中VA、VD、VE的测定,减少了前处理过程,提高了实验效率。但是,由于VE四个异构体及VD2、VD3分离难度大,目前,文献报道的多为同时检测VA、VD2、VD3、α-生育酚、β-生育酚、γ-生育酚和δ-生育酚中的其中几个,没有同时检测7种组分的方法。本实验基于GB 5009.82—2016样品前处理方法,建立一种高效、准确同时测定保健食品中7种VA、VD、VE组分的二维反相高效液相色谱法,以期为保健品中多种维生素同时检测提供一定的参考依据。
1 材料与方法
1.1 材料与试剂
视黄醇(纯度95%)、γ-生育酚(纯度96%)、δ-生育酚(纯度90%) 美国Sigma公司;α-生育酚(纯度100%) 美国Supelco公司;β-生育酚(纯度98%) 百灵威科技有限公司;VD2(纯度99.9%)、VD3(纯度99%) 中国食品药品检定研究院。
甲醇、乙腈(均为色谱纯) 德国Meker公司;无水乙醇、氢氧化钾、2,6-二叔丁基对甲酚(2,6-di-tert-butyl-4-methylphenol,BHT)、抗坏血酸、石油醚(30~60 ℃)、乙醚(分析纯) 国药集团化学试剂有限公司。
1.2 仪器与设备
1260高效液相色谱系统(配有双四元泵、自动进样器、柱温箱内置两位六通阀、2个二极管阵列检测器和Open LAB CDS数据处理系统) 美国安捷伦公司;XPE 205电子天平 瑞士梅特勒公司;Hei-VAP旋转蒸 发仪 德国Heidolph公司;Genpure超纯水仪 美国热电公司。
1.3 方法
1.3.1 色谱条件
采用中心切割的柱切换法对保健食品中VA、VD、VE进行分析,该分析过程包含3个阶段:第1阶段(0~5.35 min)为进样、检测VA,此时阀的位置为1-2;第2阶段(5.35~5.80 min)为净化、捕获VD,此时阀的位置为1-6;第3阶段(5.80~15.0 min)为第一维继续检测4种VE,第二维检测VD,此时阀的位置为1-2。系统流路见图1。
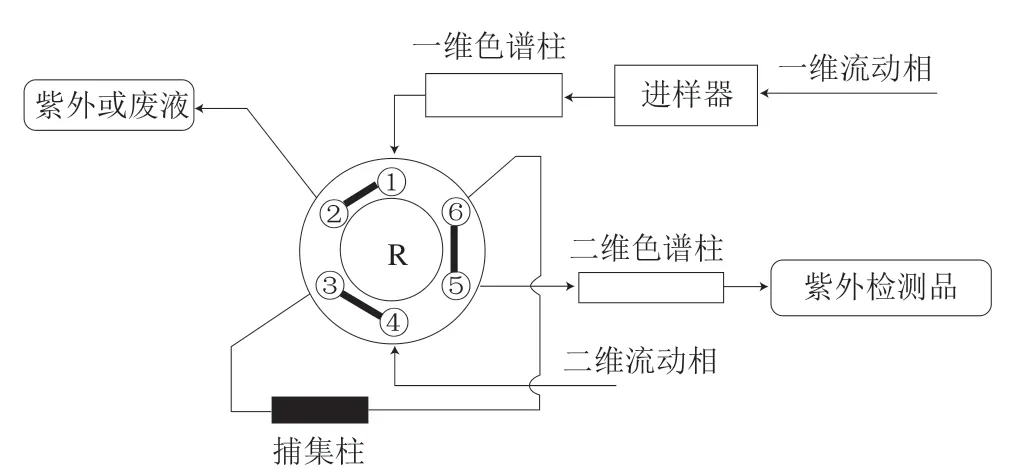
图1 二维柱切换流路系统示意图Fig. 1 Schematic diagram of two-dimensional column switching system
一维色谱柱:Agilent Poroshell 120 PFP(4.6 mm×100 mm,2.7 μm);二维色谱柱:Agilent Zorbax Eclipse PAH(2.1 mm×100 mm,3.5 μm);捕获柱:Agilent Poroshell 120 EC C18(4.6 mm×5 mm,4 μm);柱温:35 ℃;进样量:10 μL;检测波长:一维检测器325 nm和294 nm,二维检测器264 nm;一维流速:1.0 mL/min,二维流速:0.3 mL/min。流动相梯度洗脱见表1。
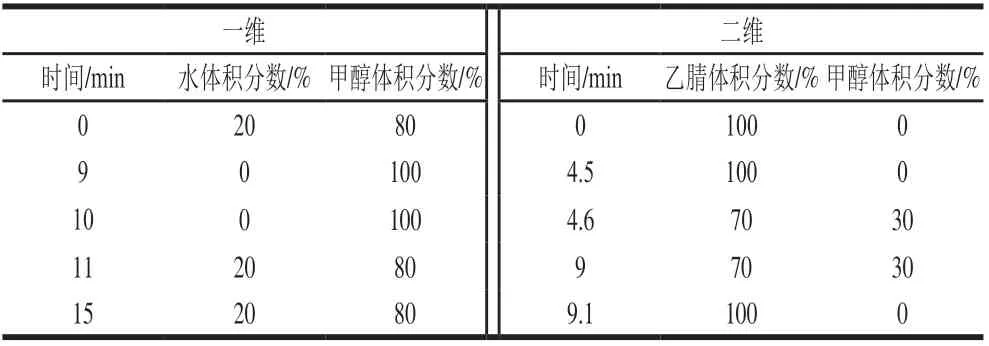
表1 梯度洗脱程序Table 1 Gradient elution procedure
1.3.2 对照品溶液的配制
分别称取视黄醇、α-生育酚、β-生育酚、γ-生育酚、δ-生育酚、VD2和VD3对照品各20 mg,于10 mL棕色容量瓶中,加无水乙醇溶解并定容至刻度,得对照品单标储备溶液。使用时用甲醇配制成系列标准混合溶液。
1.3.3 供试品溶液制备及测定
参照GB 5009.82—2016中有关样品前处理方法,样品先经过热皂化,皂化液经石油醚-乙醚 (1∶1,V/V)混合液萃取,石油醚-乙醚混合萃取液经过无水硫酸钠脱水后低温减压回收溶剂,再以甲醇溶解残留物并定容后,上机分析。本法使用的所有器皿不得含有氧化性物质;分液漏斗活塞玻璃表面不得涂油;处理过程应避免紫外光照,尽可能避光操作;提取过程应在通风柜中操作。具体操作如下:
称取2 g样品经均质后于150 mL平底烧瓶中,加入20~30 mL温水(40~50 ℃),1.0 g抗坏血酸和0.1 g BHT,混匀。加入30 mL无水乙醇,加入10~20 mL质量浓度为50 g/100 mL的氢氧化钾溶液,边加边振摇,混匀后于恒温振荡器上80 ℃皂化30 min,皂化后立即用冷水冷却至室温。一般皂化时间为30 min,如皂化液冷却后,液面有浮油,需要加入适量上述氢氧化钾与无水乙醇溶液,并适当延长皂化时间。将皂化液用30 mL水转入250 mL的分液漏斗中,加入50 mL石油醚-乙醚混合溶液振荡萃取5 min,将下层溶液转移至另一250 mL的分液漏斗中,加入50 mL的石油醚-乙醚混合溶液再次萃取,合并醚层。用约150 mL水洗涤醚层,约需重复3 次,直至将醚层洗至中性(可用pH试纸检测下层溶液pH值),去除下层水相。将洗涤后的醚层经无水硫酸钠(约3 g)滤入250 mL旋转蒸发瓶或氮气浓缩管中,用约15 mL石油醚-乙醚混合溶液冲洗分液漏斗及无水硫酸钠2 次,并入蒸发瓶内,并将其接在旋转蒸发器或气体浓缩仪上,于40 ℃水浴中减压蒸馏或气流浓缩,待瓶中醚剩下约2 mL时,取下蒸发瓶,立即用氮气吹至近干,用甲醇分次转移到容量瓶中,并定容至刻度。溶液过0.22 μm有机系滤膜后上机测定。定容体积视待测物目标浓度而定,处在线性范围内进行定量。分别精密吸取对照品溶液10 μL与供试品溶液10 μL,注入高效液相色谱仪 进行测定。
2 结果与分析
2.1 色谱柱的选择
一维色谱柱的选择主要依据其对VA与4种生育酚的分离效果。其中,4种生育酚的分离较难,特别是β-生育酚和γ-生育酚的分离对色谱柱要求较高。作为异构体的4种生育酚,普通C18柱达不到较好的分离效果。国标方法GB 5009.82—2016第一法选择C30柱分离含有4种生育酚的保健食品,且温度控制在(20±2)℃,条件较为严格,分析时间较长。Agilent Poroshell 120 PFP柱为五氟苯基柱,可用于卤代化合物的位置异构体和非卤代化合物的选择性分析,如含羟基、羧基、硝基和其他极性基团的极性化合物。且当官能团处于芳香或其他刚性环状系统上时,色谱柱选择性会增强[26]。生育酚是色满(苯并二氢呋喃)的衍生物,由一个具有氧化活性的6-羟色环和一个类异戊二烯侧链构成。根据苯环上甲基数及位置的不同分为α-、β-、γ-和δ-生育酚(图2)[27]。因此,4种生育酚可以在Agilent Poroshell 120 PFP柱上得到有效分离。
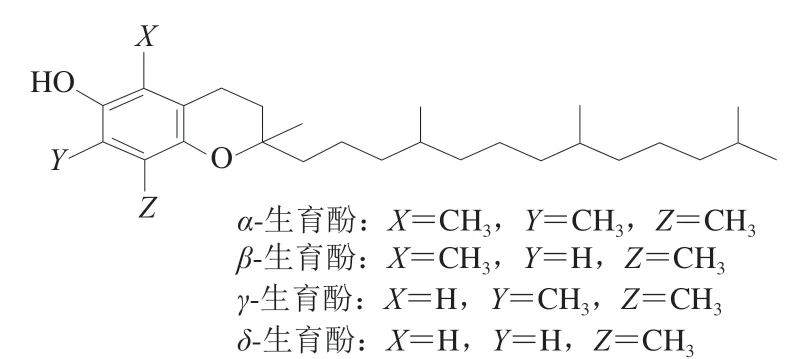
图2 生育酚的化学结构式Fig. 2 Structural formula of vitamin E
二维色谱柱的选择主要依据其对VD2和VD3的分离效果。Agilent Zorbax Eclipse PAH多环芳烃柱采用聚合C18键合填料填充,能够为多环芳烃的分离提供高选择性。其还可以用于需要空间选择性的键合相或不同键合方式的C18柱以实现分离的化合物。VD2和VD3是两种同效的维生素,其结构来源于类固醇的环戊氢烯菲环结构[28]。VD2和VD3只是侧链结构的不同,前者较后者在侧链上多了1个双键和1个甲基[29]。因此,VD2和VD3可以在Agilent Zorbax Eclipse PAH柱上得到有效分离。
二维液相色谱条件选择需考虑色谱柱的差异性和流动相的兼容性[12]。Agilent Poroshell 120 PFP (4.6 mm×100 mm,2.7 μm)柱和Agilent Zorbax Eclipse PAH(2.1 mm×100 mm,3.5 μm)柱良好的正交性和流动相的相容性,确保了4种生育酚的有效分离及VD2和VD3的准确定量(图3)。
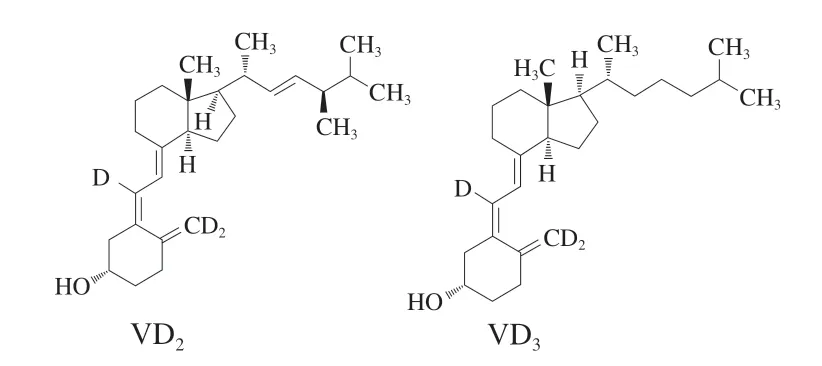
图3 VD的化学结构式Fig. 3 Structural formula of vitamin D
2.2 阀切换时间的确定
采用在线柱切换二维色谱法分离样品中的VA、VD、VE。样品进样后通过一维色谱柱进行初步分离,通过阀切换将含有VD2和VD3的馏分切割至捕获柱,捕获柱与二维色谱柱串联,由二维色谱柱进一步分离VD2和VD3,一维色谱柱继续分离4种生育酚,一维检测器检测VA与生育酚。二维检测器检测VD2和VD3。阀切换图见图1。为确定阀的准确切换时间,减少溶剂效应和提高回收率,需要确定VD2和VD3出峰时间的起点和终点。通过关闭阀切换命令在一维色谱柱与一维检测器上进行标准品分析,测定VD2和VD3的出峰时间起点和终点。由于使用捕获柱收集一维色谱柱的VD2和VD3的馏分,而捕获柱柱容量小,需要考虑在阀切换的时间段内捕获柱是否能完全将VD2和VD3收集[16]。因此,需要在一维色谱柱上尽量减小VD2和VD3的分离度,缩短峰宽。通过多次实验确定,Agilent Poroshell 120 PFP(4.6 mm×100 mm,2.7 μm)为一维色谱柱,流速为1.0 mL/min,由标准品测试确定出阀切换时间为5.35 min,更改阀的位置端口1-6,5.80 min更改阀的位置端口1-2。
2.3 取样量的选择
目前,食品中VA、VD、VE的测定主要依据GB 5009.82—2016,固体样品取样量为2~5 g或5~10 g,取样量较大且不固定,这主要是因为食品种类繁多,剂型不固定,含量差距较大。保健食品相对食品有较固定的剂型,且保健食品主要是以补充营养物质为目的,而人体需要的维生素含量有一定的范围,所以市售保健食品中维生素含量有较固定的范围。本研究调研了国内几大维生素生产企业市售VA、VD、VE类保健食品,结果显示,VA、VD、VE类保健食品主要以片剂和胶囊剂为主,VA含量范围为0.1~1.0 mg/g,VD含量范围为0.69~17.2 μg/g,VE含量范围为3.7~500 mg/g。其中,单方VE保健食品VE含量范围为80~500 mg/g,复方VE保健食品VE含量范围为3.7~26.2 mg/g。因保健食品样品较均匀,且较大取样量可能对提取效率产生影响,结合VD较低的含量,选择2 g作为样品取样量,10 mL作为测定VD时的定容体积,VA、VE稀释至线性范围内进行定量。
2.4 提取溶剂的选择
选择石油醚-乙醚混合液萃取保健食品中VA、VD、VE。因乙醚为易制毒试剂,使用时受到严格管制,为方便实验,考察只用石油醚的萃取效果。结果表明,石油醚提取只含有VA、VD和α-生育酚的保健食品时萃取效果与石油醚-乙醚混合液没有显著差异;萃取含有β-生育酚、γ-生育酚、δ-生育酚3种生育酚的保健食品时,萃取效果不好,这是因为这3种生育酚较α-生育酚极性强,根据相似相溶原理,弱极性的石油醚对他们达不到较好的萃取效果。故在提取含有这3种生育酚的保健食品时必须用石油醚-乙醚混合液。
2.5 标准曲线与定量限测定
精密吸取各对照品单标储备溶液,加甲醇配制成VA质量浓度为2.50、10.0、20.0、40.0、50.0 μg/mL,4种生育酚质量浓度为 5.00、20.0、40.0、80.0、100.0 μg/mL, VD2和VD3质量浓度为0.10、0.20、0.50、1.00、 2.00 μg/mL的标准系列混合溶液。分别吸取系列标准混合溶液各10 μL,依次注入液相色谱仪。以质量浓度(μg/mL) 为横坐标,峰面积为纵坐标,计算回归方程。以10 倍信噪比确定定量限。结果(表2)表明:VA在2.50~50.0 μg/mL线性范围内,4种生育酚在5.00~100.0 μg/mL线性范围内,VD2和VD3在0.10~2.00 μg/mL线性范围内,线性相关性良好。
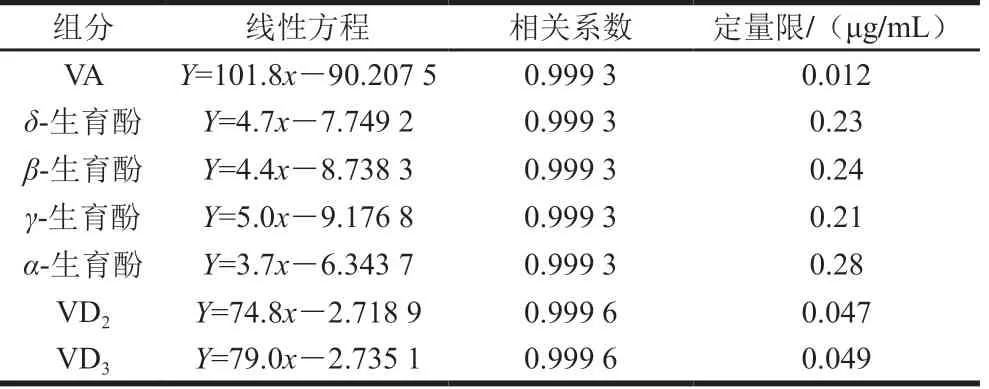
表2 7种化合物的线性方程、相关系数和定量限Table 2 Linear equations, correlation coefficients and limits of quantitation for seven vitamin compounds
2.6 精密度及系统适用性检测
分别精密吸取VA质量浓度为50.0 μg/mL、4种生育酚质量浓度为100.0 μg/mL、VD2和VD3质量浓度为2.00 μg/mL的标准混合溶液10 μL,连续进样6 次,记录峰面积。相对标准偏差(relative standard deviation,RSD)均小于0.5%。各组分理论塔板数均大于10 000,分离度均大于1.5(表3)。表明仪器精密度良好,满足系统适用性要求。标准混合溶液图谱见图4。
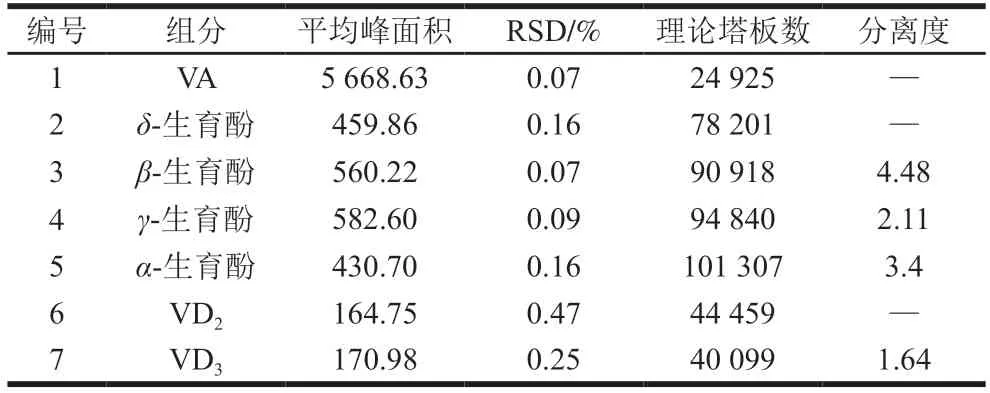
表3 精密度及系统适用性结果Table 3 Results of precision and system suitability

图4 标准品二维液相色谱图Fig. 4 Two-dimensional chromatogram of mixed vitamin standard solution
2.7 回收率与重复性实验结果
采用阴性样品加标回收法测定回收率。分别向阴性样品中加入适量对照品溶液,按1.3.3节方法制备供试品溶液,使加标溶液质量浓度分别为VA:5.0、20.0、40.0 μg/mL;4种生育酚:10.0、40.0、80.0 μg/mL;VD2和VD3:0.20、0.80、1.60 μg/mL,分别作为低、中、高3个水平加标溶液。平行制备3 份样,计算回收率,作为回收率样品。中水平加标溶液平行制备6 份样,作为重复性样品,记录峰面积,计算RSD。不同质量浓度水平平均回收率为89%~99%,RSD均小于4%。表明该方法准确度高、重复性好,结果见表4、5。

表4 样品加标回收率及RSDTable 4 Recoveries and RSDs for spiked samples

表5 重复性结果Table 5 Results of repeatability
2.8 样品测定结果
选取市售VA、VD、VE类保健食品,采用本法对样品进行检测。检测结果发现,市售VD类保健食品中主要添加VD3,VE类保健食品中主要添加的为α-生育酚,所测样品均符合各企业标准规定,结果见表6,某样品图谱见图5。
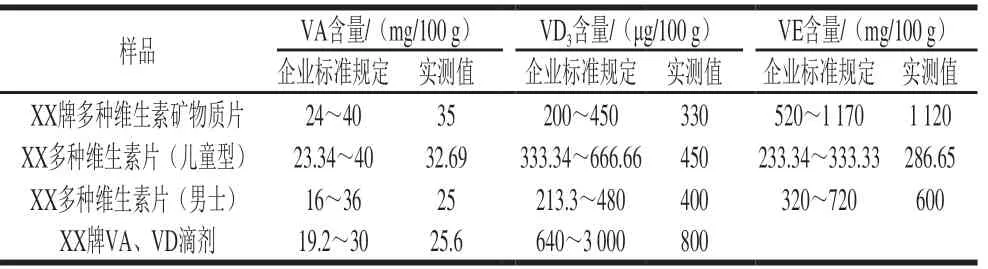
表6 样品测定结果Table 6 Results of vitamin determination in actual samples

图5 样品二维液相色谱图Fig. 5 Two-dimensional chromatograms of samples
3 结 论
本实验基于在线柱切换法二维液相色谱技术建立了保健食品中VA、VD、VE的测定方法,系统适用性实验结果表明,本方法可满足VA、VD、VE的测定要求,利用一维色谱柱可完成VA、4种生育酚的分离和VD的净化,利用二维色谱柱可完成VD2、VD3的分离及定量分析。本方法的线性关系、准确度和精密度良好,采用优化的在线二维系统连接设计,一次进样可完成VA、VD、VE的定量测定,大大提高了实际样品的分析效率,可进行推广应用。
