氧气在碳化钨负载的铜-金合金单层上吸附与解离的第一性原理研究
2022-03-04常青芳张喜林
常青芳,张喜林
( 河南师范大学 物理学院 河南省光伏材料重点实验室,新乡 453000)
1 引 言
氧气分子在催化基底上的吸附、解离是很多重要反应,如氧气还原反应( ORR)[1-4],一氧化碳氧化[5-7]等的关键步骤.通常来说,过强的吸附利于氧气的解离,但较强的氧原子吸附会限制其进一步反应; 太弱的吸附则会抑制解离的发生,阻止氧原子生成和进一步参与反应; 而温和的吸附既有利于氧气的解离又不至于引起催化剂的氧化失活[8,9].已有研究表明,反应气体的吸附强度与催化剂的电子结构之间存在直接联系[10-12].以金属催化剂为例,一般而言,金属催化剂的d电子中心越靠近费米能级,对反应物的吸附愈强;反之则吸附越弱[13].因此,调节金属催化剂的电子结构对实现氧气分子的中等吸附,及氧气解离势垒和氧原子进一步反应的平衡至关重要.
合理设计负载型催化剂可以有效调节负载催化剂的电子结构,调控氧气分子的吸附与解离.我们先前的研究表明,碳化钨作为基底可以显著调节负载金属单层催化剂的电子结构,致使氧气的吸附构型、吸附能及解离势垒与纯金属相比有很大差异[14,15].同时,不同的金属单层在碳化钨上也具有各自独特的电子结构和氧气吸附特征.具体来说,碳化钨负载的钯单层和铂单层的d 带中心更加靠近费米能级,氧气分子的吸附能也因此比在金单层大了约1 eV,分解势垒则降低了约0.5 eV.计算结果也表明碳化钨负载的铂钯金单层并不能有效的促进氧气的解离,特别是在负载的金单层上,较弱的氧气吸附反而不利于其分解[15].系统的反应过程和机理研究表明,弱的氧气吸附促使氧还原反应沿着连续加氢的路径进行,从而打破氧分子双键.但在一定反应条件下,较弱的吸附需要更高的氧分压才能使反应正向进行.通过进一步地表面调控,使催化剂对氧气分子的吸附强度更加温和,从而加速氧气的分解,同时保持快速的氧原子反应,对设计氧化还原反应大有裨益.
表面合金化是将两种或多种不同的金属原子组合形成.合金化元素由于原子半径大小不同,将不可避免的引入表面应力、掺杂等效应,从而引起表面几何结构与电子结构的差异,致使反应物吸附构型和能量的改变[16-19].碳化钨负载的贵金属钯金合金单层已被实验合成,并且展现出了卓越的催化活性、循环稳定性和抗毒性[20].理论上,赵等人[21]发现在碳化钨负载的银单层上掺入微量的钯可以提升合金表面对氧分子的捕获能力,同时不会对一氧化碳催化氧化的性能产生较大影响,研究结果初步说明了表面合金化对氧气吸附的促进作用.碳化钨负载的金单层在几何结构和电子特性上具有和银单层相似的性质[22],对金单层进行合金化,研究合金化对氧气吸附和解离的影响,并和银单层的研究结果作对比,揭示合金化活化分解氧气的机制及影响氧原子吸附的原理,为深入氧化还原反应的内在本质提供理论基础.
我们采用基于密度泛函理论的第一性原理方法,结合电荷布局分析、过渡态计算等研究铜掺杂的金单层对氧气分子吸附、解离的影响和促进机制.分析不同的铜掺杂构型和浓度的结构稳定性、电子特性及其对氧分子解离与氧原子吸附的影响.并与已有的结果做深入对比,揭示合金单层在催化氧气解离方面的优势.本研究工作将对包含氧解离过程的催化反应有参考意义,同时有利于进一步设计高效廉价的氧化还原催化剂.
2 计算方法与模型
自旋极化总能计算均采用Material studio 中的DMol3模块完成[23,24].采用广义梯度近似下的PW91 泛函用来描述交换关联函数[25-27],离子和电子间的相互作用用DFT 半核心赝势方法来描述[28].截断基组设置为4.4 Å.几何优化能量的收敛标准设置为10-6Ha,原子力的收敛判据为0.002 Ha/Å,位移设置为0.005 Å.电子自洽精度设置为10-6Ha.布里渊积分采用Monkhorst-Pack形式的特殊K 点产生方法[29],设置5 × 5 × 1 的K 网格点.过渡态计算( TS) 采用完全线性同步输出( LST) /二次同步输出( QST) 方法.
吸附能定义为
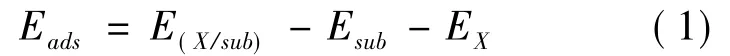
其中E(X/sub)和Esub分别代表有和没有吸附物的基底的最稳定构型的总能量,EX是基态下相应的分离吸附物的总能量.较小的Eads值意味着较强的吸附.负值表明放热的吸附过程.
负载单层的表面能定义[30]为

其中EMML/WC和EWC分别代表有与没有负载单层的碳化钨的总能量,ECu代表单个铜原子总能量,EAu为单个金原子总能量,n 为铜原子个数.Ef值越负,该单层就越容易形成.
文献研究表明金属单层在以钨为终止面的碳化钨表面的稳定性比在以碳为终止面的碳化钨表面更强[31].因此本研究选择以钨为终止面的碳化钨[0001]表面( WC( 0001) ) 为基底,构建了包含三个WC 双层的(3 ×3) 的超胞.为避免镜像相互作用,在垂直方向上设置了18 Å 的真空层.优化WC 体块得到的晶格常数为2.920 Å,与文献相当[15,31],说明我们计算参数的合理性.为了模拟体块的效果,我们固定了底部的4 层原子,剩余部分在结构优化过程中自由弛豫.我们计算并对比了铜、金原子层在碳化钨表面不同位置的总能量,计算结果表明,金属原子吸附在WC(0001) 的hollow 空位上( 次表面碳原子的正上方)具有较低的总能量.这与先前的理论计算相一致[15],因此本文的研究均基于此类结构展开.
根据氧气在纯金属单层上的可能吸附位置,我们选择将金单层中一个或两个邻近的金原子替换为铜原子,结构俯视图和侧视图均展示在图1( b,e,c,f) 中.同时考虑了间隔位置掺杂,结果显示其对氧气的吸附与单个铜原子掺杂一致,所以不再在文中讨论.金单层在碳化钨上负载已经被实验合成( AuML/WC( 0001) )[32],第一性原理能量学和分子动力学研究也证实了其在反应条件下的稳定性[31].为分析掺杂铜原子后的结构稳定性,我们计算了表面合金结构的表面能,并与纯的金单层作对比.发现掺杂铜后合金单层的表面能更负,由纯Au 单层的 -3.65 eV,降低到Cu1Au8/WC( 0001) 的 - 3.70 eV 及Cu2Au7/WC(0001) 的 -3.75 eV.表面能的降低主要是由于合金单层中金原子与铜原子之间及他们与碳化钨( WC) 衬底之间增强的相互作用引起的.这种现象与金属原子之间的键长( d(Cu-Au)) 及金属原子与碳化钨( WC) 表面沿竖直方向的垂直距离缩短相一致.这些结果表明,铜掺杂有助于增强合金催化剂的稳定性.
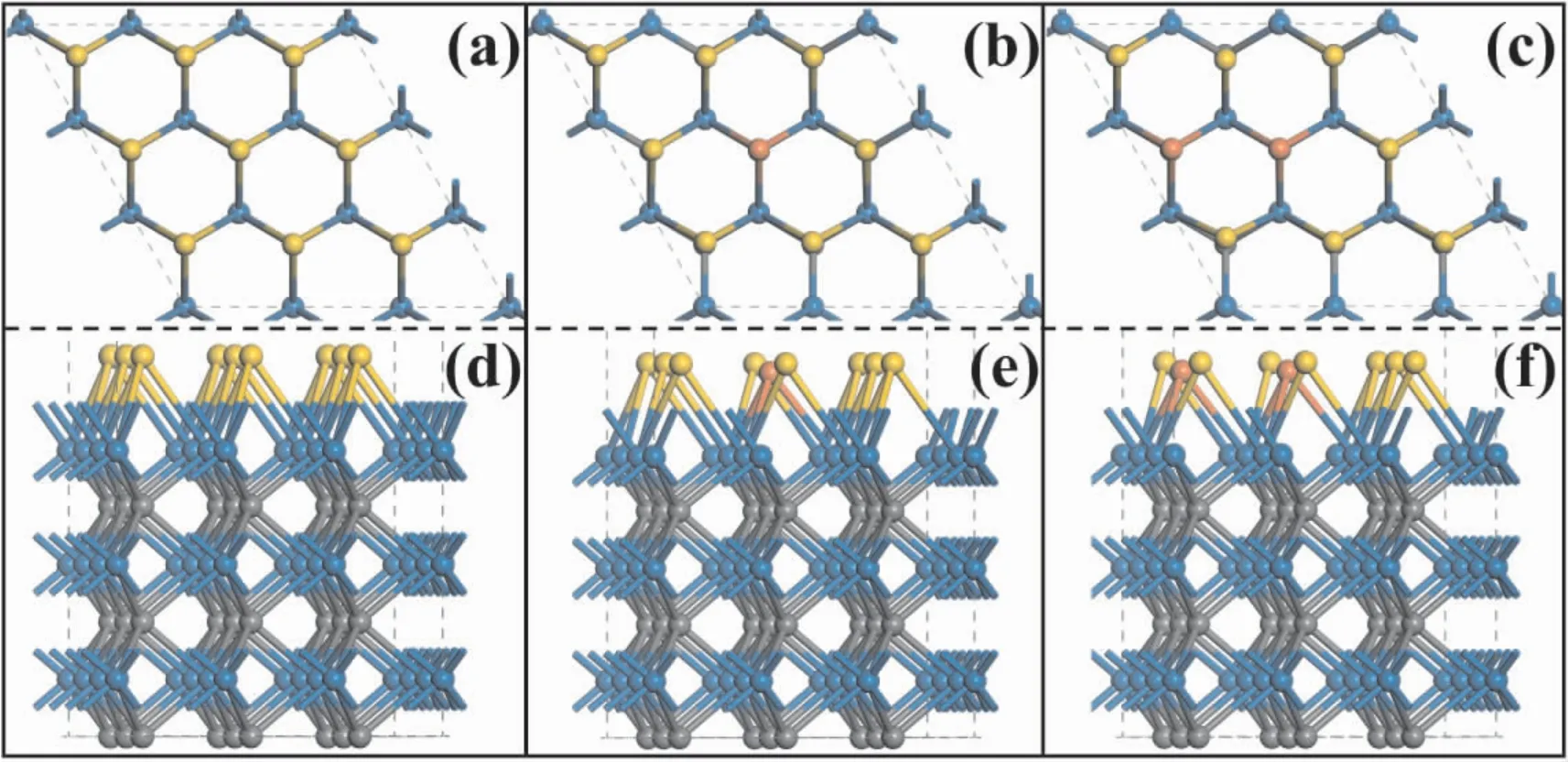
图1 碳化钨负载的纯金单层、掺杂一个和两个铜原子的铜金合金单层的俯视图( a -c) 与侧视图( d -f).其中,黄色、橙红色、蓝色和深灰色分别代表金、铜、钨和碳原子.Fig.1 The top views ( a-c) and side views ( d-f) of pure Au monolayer and Au monolayer doped with one or two Cu atoms supported on tungsten carbide.While yellow,orange,blue and dark gray represent Au,Cu,W and C atoms,respectively.
3 结果与讨论
3.1 O2分子在MML/WC(0001)表面的吸附
文献中AuML/WC(0001) 催化氧气分子解离的势垒高达1.56 eV,主要是由于氧气在金单层上的吸附较弱[15].当前研究尝试通过掺杂电负性较强的铜原子,调节表面合金的电子结构,增强单层对O2的吸附,从而促进氧气解离.我们首先对碳化钨( WC) 负载的纯金单层,掺杂一个和两个铜原子的金单层( MML/WC(0001) ,M=Au,Cu -Au) 进行了电荷布局分析研究.如图2( a) 所示,在纯金单层中每个金原子均得到0.205 个电荷.掺杂一个铜原子后,铜原子向衬底及周围的金原子转移0.749 个电荷,铜原子周围的金原子均得到了更多的电荷( 约0.057).掺杂铜原子达到两个后,与铜相邻的金原子得电子数再次增加,铜原子失电子数减少,均为0.685.这些结果表明铜原子掺杂会调节表面电荷的重新分布,创造新的可能的吸附位点,改善氧气分子及氧原子的吸附与反应性质.
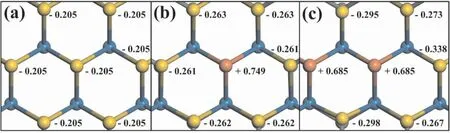
图2 碳化钨负载的纯金单层及有一个和两个铜原子掺杂的合金单层的电荷分布.Fig.2 The charge distributions of pure gold monolayer and gold monolayer doped with one and two cooper atoms supported on tungsten carbide.
为验证以上推断,我们首先研究了氧气分子在三种表面上的吸附特点.如图3( a) 所示,O2分子在纯金表面稳定吸附于两个相邻金原子的桥位,吸附能为- 0.17 eV.吸附后的O2分子键长由1.23 Å 拉伸至1.35 Å.Cu1Au8/WC(0001) 体系中氧气倾向于吸附在铜的顶位与相邻hcp 空位的连接处( thb) ,吸附能相比于纯金体系增强了0.33 eV,同时相应的O - O 键长也增长了0.04 Å.Cu2Au7/WC(0001)体系中,氧气分子斜跨过Cu-Cu桥位,两个氧原子分别靠近于fcc 空位和Cu -Au桥位,吸附能和O-O 键长分别增加到- 0.93 eV和1.45 Å,与在Au38团簇上H2O 与O2分子共存时的吸附强度相当[33].这些结果说明在Cu2Au7/WC(0001) 表面,氧气解离更容易发生.
诸多反应中,孤立氧原子的吸附也至关重要.图3( d) 中可见孤立O 原子倾向于吸附在三个金原子的fcc 位,吸附能为3.37 eV.而在金单层掺杂一个铜原子后( 图3( e) ) ,氧原子的吸附能增加了0.49 eV,最稳定吸附位置为铜原子与金原子形成的fcc 位点.当替换两个金为铜原子后,氧原子的吸附强度为4.39 eV,与文献中氧原子在PdML/WC( 0001) 和PtML/WC( 0001) 上吸附能接近[15].
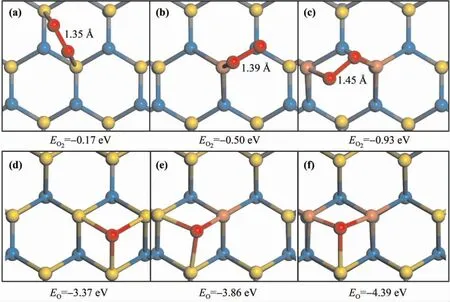
图3 O2分子( a -c) 及孤立的O 原子( d -f) 在AuML/WC( 0001) ,Cu1 Au8/WC( 0001) 和Cu2 Au7/WC(0001) 表面的最稳定吸附构型及相应吸附能,氧气吸附后的O-O 键长也在图中标注.Fig.3 The most stable configurations and corresponding adsorption energies of oxygen molecule and isolated oxygen atom adsorbed on AuML/WC(0001) ,Cu1Au8/WC(0001) and Cu2Au7/WC(0001).The O -O bond length of adsorbed oxygen is also labeled.
3.2 O2在MML/WC(0001)表面的解离
我们进一步研究了合金表面上氧气的解离过程.在Cu1Au8/WC(0001) 表面,2 个氧原子最稳定吸附构型为两个氧原子分别吸附在两个金原子和一个铜原子形成的fcc 位和hcp 位( 图4a,FS).在Cu2Au7/WC(0001) 表面,两个氧原子分别稳定吸附在两个铜原子两侧的Cu1Au2fcc 位和Cu2Au1fcc 位( 图4b,FS).以氧气在两种合金表面最稳定吸附构型为初始结构( IS) ,通过过渡态搜索得到氧气分子解离的最佳路径.如图4a 所示,单个铜原子掺杂时,初态( IS) 时氧分子稳定吸附在thb位置,随后Cu -top 位的O 原子跨过铜原子,向两个金原子和一个铜原子形成的fcc 位移动,而另一个氧原子则在hcp 位附近弛豫.整个过程共释放了0.40 eV 的能量,需要克服1.28 eV 的过渡态势垒.此时,两个氧原子的距离由初态的1.39 Å 拉伸至3.13 Å,解离为两个孤立的O 原子.图4b 为O2在Cu2Au7/WC(0001) 体系上的解离过程.如图中所示,氧分子由初态解离为吸附在fcc 空位的两个氧原子,仅需克服0.64 eV 的势垒,同时释放出0.54 eV 的热量.这些结果表明铜原子掺杂会改变O2的吸附构型,增大氧分子的吸附能,减小氧气分子的解离势垒,可能促进氧还原反应或一氧化碳反应的发生.
为深入探究产生此效应的物理本质,我们计算对比了三种体系的态密度.如图5 所示,铜掺杂会引入新的态密度峰.这些Cu -d 态峰相对于Au-d 态更加靠近费米能级,意味着铜可能是潜在的活性位点,具有更强的分子吸附能力.这与前边的吸附研究结果一致.随着铜原子掺杂个数增加( a 到c) ,O2在费米能级处的反键峰逐渐降低并下移,表明其氧分子得到更多的电子,O -O键能逐渐减弱,这与逐渐增大的O-O 键长(1.35 Å <1.39 Å <1.45 Å) 相吻合.对比5b 和5c 费米能级附近Cu-d 和O2态密度曲线,可以发现掺杂两个铜原子时Cu -d 态与O2分子有更明显的叠加,说明他们之间有更强的作用力,从而导致O2更容易解离.

图5 有O2分子吸附的AuML/WC(0001) ,Cu1Au8/WC(0001) 以及Cu2Au7/WC(0001) ( 对应图3 中a-c) 表面金属( M) 及吸附的氧气分子的态密度曲线.其中Au-d,Cu-d 均为平均值.Fig.5 Density of states of metals and adsorbed O2 on the surfaces of AuML/WC(0001) ,Cu1 Au8/WC(0001) and Cu2 Au7/WC(0001) ,respectively.While Au-d and Cu-d are averaged.
4 结 论
上述结果表明碳化钨负载表面合金在催化氧反应方面具有一定的优越性.我们总结对比了氧气在其他催化剂表面吸附及解离的性能,具体数据如表1 所示.与贵金属铂和钯相比,碳化钨负载铜-金合金对氧气分子和氧原子的吸附更强,氧气解离更容易.同时,相对较弱的氧原子吸附能也有助于合金单层催化剂的复原,提高催化活性.在碳化钨负载的铂和钯单层比碳化钨负载的铜-金合金单层对氧分子的吸附较强,但氧解离势垒并没有降低.这主要由金属d 态与氧气2π反键态相互作用不同导致的.相比于金表面,氧气在碳化钨负载的金单层上的吸附虽略有增加,但活化势垒仍然较大.通过适量的铜原子掺杂,碳化钨负载的金单层对氧气和氧原子的吸附得到增强,氧气解离势垒从1.56 eV 下降到0.64 eV.当前的结果证明表面合金化引起的配位效应及局部应力能够促进电子结构重排,改变催化剂对吸附物的吸附强度,从而提高催化剂活性.充分利用表面合金效应及基底与负载型催化剂强相互作用,有助于设计更加高效、稳定、廉价的催化剂.

表1 不同的催化剂表面氧气吸附时的O-O 键长、吸附能,氧原子的吸附能及氧气解离的过渡态势垒和能量变化.Table 1 The bond lengths of O-O,adsorption energies of oxygen ( Eads( O2) ) and isolated oxygen atom ( Eads( O) ) ,the transition state energy barriers( Ea) and energy changes ( ΔE) of oxygen dissociation on different catalyst surfaces.
