Nogo-B 与JCAD 诱导肝癌基因激活
2022-03-04郭阵雨赵新军马军仁
郭阵雨,赵新军,2,马军仁
(1.伊犁师范大学新疆凝聚态相变与微结构实验室,伊宁 835000;2.伊犁师范大学 微纳电传感器技术与仿生器械实验室,伊宁 835000)
1 引 言
慢性肝脏炎症会诱发肝癌( HCC) ,例如: 如慢性乙型肝炎( HBV) 或丙型肝炎( HCV) 、非酒精性脂肪肝炎( NASH) 会导致肝炎转化为HCC.感染HBV 或HCV 病毒后会造成肝细胞基因改变,从而导致HCC 的发生发展.NASH 临床表现为代谢综合征,生物学和流行病学研究表明,NASH可进展为HCC 或其他晚期肝病[1,2].
目前的研究已经发现,在NASH -HCC 转变过程中,脂肪变性发展和自噬功能降低均与NASH 有关[3-5],炎症和内质网应激则会在很大程度上影响NASH 的发病.近年的研究[6]也初步确认了patatin - like phospholipase domain containing 3( PNPLA3) 基因、transmembrane 6 superfamily member 2( TM6SF2) 基因和其他致病基因的功能障碍与NASH 有关,尽管这些基因的功能尚未完全确定,但是发现其多态性与HCC 的发生有关.NASH 发病的多因素特征及其进展决定了脂肪变性微环境中NASH - HCC 发生的致病复杂性,Wolf 等人[7]通过基因敲除操作,研究了小鼠细胞的适应免疫性在NASH - HCC 转化过程中的作用,该研究表明抑制Th17 细胞分化或阻断IL -17A 信号传导可阻止NASH 和随后的HCC 发生.Yoshimoto 等人[8]的研究证明了肠道菌群在NASH进展中的重要作用,并发现衰老的肝星状细胞( HSC) 对于将脂肪变性肝细胞转化为恶性细胞是至关重要的.在NASH—肝癌的发展过程中,脂代谢异常是NASH 转变为肝癌的一个重要促成因素,脂质的分解不仅为细胞提供能量,而且还调节其他的细胞过程,如激活致癌信号通路.Zhu等人[9]研究发现,内质网定位蛋白Nogo -B 的缺失会减少原发HCC 中STAT3 的磷酸化,这样Nogo-B 可以通过调节IL -6/STAT3 信号影响HCC 的发生.最近,Tian 等人[10]的实验结果表明,在NASH 向HCC 转化过程中,Low density lipoprotein ( LDL) 蛋白先被氧化为oxidized low density lipoprotein ( oxLDL) ,oxLDL 会诱导Nogo-B 表达上调,进而提升Hippo 信号通路中Yes-associated protein ( YAP) 的活性,激活HCC 基因的表达.Ye 等人[11]的研究则发现,在NASH所致HCC 的进展过程中,冠状动脉疾病相关连接蛋白JCAD 通过抑制Hippo 信号通路中large tumor suppressor homolog 2 ( LATS2) 激酶,抑制YAP 蛋白磷酸化,促进NASH 发展为HCC.深刻理解NASH 诱发HCC 发生发展的信号通路特性,对于设计阻断NASH 诱发的HCC 治疗方案是非常必要的.在激活基因通路研究方面,之前的理论结果[12-16]已经定量揭示了肿瘤抑制的物理机制.在慢性NASH 微环境中,oxLDL 通过诱导Nogo-B 表达上调提高YAP 的活性,激活HCC 基因的表达[10].同时,JCAD 通过抑制Hippo 信号通路中的LATS2 提升YAP 的活性,促进HCC 的发生发展[11].与JCAD 调控效应相反,LATS2 则促进YAP 磷酸化,限制其活性,进而降低了connective tissue growth factor ( CTGF) 等增殖因子的表达,抑制HCC 细胞生长增殖[11,17].那么,Nogo-B、JCAD 与LATS2 是如何协同调控NASH-HCC的发生发展,当前仍然没有深刻理解.
本文建立理论模型研究Nogo -B 诱导oxLDL降解,以及JCAD 与LATS2 相互作用调节YAP磷酸化,协同调控HCC 基因激活的动力学机制.分析Nogo - B 诱导的oxLDL 降解、JCAD 与LATS2 调节YAP 磷酸化,激活致癌因子的信号通路特性,进一步深刻揭示NASH 诱发HCC 的致癌机理.
2 理论模型
在NASH 所致的HCC 进展过程中,我们考虑: (1) Nogo-B 通过激活DoxLDL -Nogo -B -YAP 通路[12],导致HCC 的发生发展; (2) JCAD与LATS2 相互作用,并抑制YAP 磷酸化,未被磷酸化的YAP 激活下游Hippo 信号通路中下游与肿瘤形成相关的基因,从而导致HCC 的发生发展.模型示意图为:
基于我们之前的研究[12],Nogo -B 与Autophagy-related 5 gene ( ATG5) 相互作用形成复合体NA,进而促进oxLDL 降解产生讲解后的ox-LDL( DoxLDL).JCAD 与LATS2 相互作用,与JCAD 结合的LATS2 标记为BLATS2,JCAD 与BLATS2 作用形成的复合体为JB,形成NA 和JB的反应均为二级反应.JB 抑制YAP 磷酸化,增进YAP 活性,磷酸化的YAP 标记为pYAP.未与JCAD 结合的LATS2 标记为FLATS2,FLATS2 则促进YAP 磷酸化,抑制YAP 活性.基于Hill 动力学与Michaelis-Menten 方程[18,19],可以获得各组分浓度随时间演化的动力学方程组为:

以上方程组中,C=[DoxLDL]+[oxLDL],表示DoxLDL 和oxLDL 的总浓度.LATS2 激酶的总浓度为[LATS2]=[FLATS2]+[BLATS2],方程(9) 中Hill 系数为n=3.累积的肝脏脂肪诱导JCAD 的表达,并与LATS2 相互作用抑制YAP 磷酸化,被磷酸化的YAP 会滞留在细胞质中,而未被磷酸化的YAP 则会转移到细胞核与TEAD 结合,激活下游的Hippo 信号通路中下游connective tissue growth factor ( CTGF) 、cysteine -rich 61 ( Cyr61) 、cyclin D1( CCND1) 和glioma -associated oncogene homologue( GLI -2) 等与细胞增殖和肿瘤形成相关的基因,从而导致HCC 的发生发展[11].在这里我们考虑CTGF 代表CYR61、CCND1 和GLI-2 等与细胞增殖和肿瘤形成相关的基因.
3 结果与讨论
Tian 等人[10]的实验发现oxLDL 降解完成需要约5 ~10 小时,DoxLDL 通过oxLDL -Nogo -B-YAP 信号通路激活下游HCC 基因.Ye 等人的实验发现[15]JCAD 通过抑制LATS2 激酶活性提升YAP 激活促进NASH 进展为HCC.为了符合实验结果,取适当的参数值( 如表1 所示) ,可以考察分析Nogo-B 与JCAD 诱导肝癌基因激活通路的动力学特性.

表1 模型中的参数取值Table 1 Standard parameter values of the model
图2a 呈现了DoxLDL、oxLDL 随时间演化的动力学关系,以及[LPC]随[DoxLDL]的变化关系.从图2a 可以看出,DoxLDL 在短时间内很快升高到了一定的浓度值,并趋于稳定,[oxLDL]则在短时间内很快降低,并趋于较低稳定值不变.由此表明,在较短的时间内oxLDL 降解,产生了大量的DoxLDL,并且在10 小时后oxLDL 降解完成,DoxLDL 维持一定的恒定值( oxLDL 已经降解完成) 不变,进而促进 Lysophosphatidylcholine( LPC) 合成,激活大量Lysopho - sphatidic acid( LPA) ( 图1a 插图) ,启动下游信号通路.从图2b 中LPC 随DoxLDL 演化的函数关系可以看出,随着[DoxLDL]的增加,[LPC]首先缓慢增加,当[DoxLDL]增加到约14 nM ,也就是oxLDL 几乎完全降解时,[LPC]出现了急剧增长.图2b 中的插入图表明[LPC]经过约20 小时升到了较大的稳定值,并开始维持这一较高的浓度值不变,反映出DoxLDL 对LPC 合成调节的时滞性.这是由于,在随着oxLDL 降解使得DoxLDL 浓度升高,一定浓度的DoxLDL 才能对LPC 合成有显著的促进作用,致使LPC 的浓度经过约20 小时很快提升,并升高到了较大的浓度值.由此表明,ox-LDL 的降解到一定程度,降解的oxLDL 达到一定的浓度值,会很快激活大量下游激活因子( autotaxin( ATX) 、LPA 等) ,之后便会在很大程度上提高Hippo 信号通路中YAP 的活性,激活下游癌基因( CTGF 等) 的表达.oxLDL 在降解为DoxLDL的过程中,Nogo-B 起着重要的调控作用[10],ox-LDL 通过CD36 大量摄入细胞内,诱导Nogo -B和CCAAT 增强子结合蛋白β ( CEBPβ) 表达上调( 如模型图1) ,在oxLDL 的降解过程中,Nogo -B 通过与ATG5 反应结合形成复合体NA,NA 调节oxLDL 的降解产生降解后的DoxLDL.另外,CEBPβ 能够增强Nogo-B 基因的转录活性,协同诱导Nogo - B 表达上调[20].为了考察NA 促进oxLDL 的降解,以及于CEBPβ 协同诱导Nogo-B表达上调效应,可以考察Nogo -B 和复合体NA浓度随时间的演化.
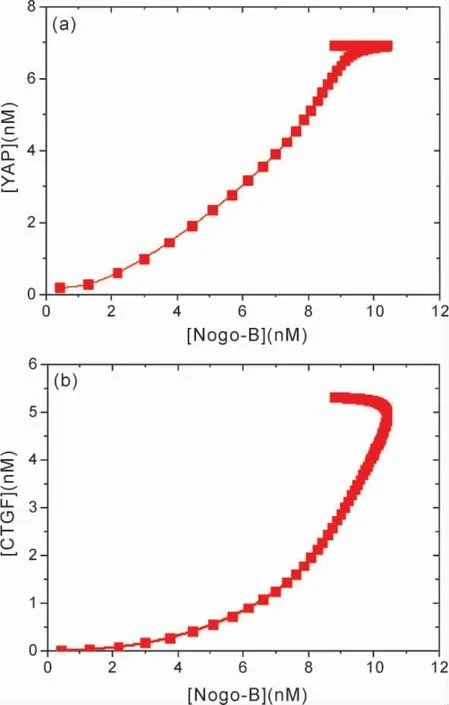
图1 Nogo-B 与JCAD 诱导肝癌基因激活的模型示意图.Fig.1 Schematic depiction of the model of Nogo -B and JCAD inducing hepatocellular carcinoma gene activation.

图2 [DoxLDL]、[oxLDL]随时间演化的动力学关系( a) ,以及[LPC]随[DoxLDL]的变化关系( b).Fig.2 Temporal evolutions of the levels of[DoxLDL]and[oxLDL]( a).The[LPC]as a function of[DoxLDL]( b).
图3a 和3b 显示了Nogo-B 与NA 浓度随时间演化的动力学关系.从图3a 可以看出,Nogo-B 经过约25 小时时间升高到了最大的浓度值,然后趋于稳定,由此表明,经过约25 小时Nogo-B的表达可以上调到最高水平,而后轻微减小并趋于稳定值不变.图3b 显示NA 经过约25 小时升高到了最大值,之后也呈现出最高表达水平而后趋于稳定值不变.比较图3a 中Nogo -B 的演化和图3b 中NA 的演化特性,可以发现,Nogo -B和NA 呈现了相似的随时间演化特性,但是NA演化幅值远大于Nogo -B 幅值.由此表明,Nogo-B 与ATG5 的结合反应决定着的NA 的合成,通过NA 的合成,ATG5 放大了Nogo -B 表达的信号,使得Nogo-B 的激活效应增强,很快提升了oxLDL 的降解( 图3b 插图) ,促进了大量的LPA 合成.这样,通过Nogo-B 增强LPA 的合成激活YAP; 当ATG5 敲减后,NA 的合成被限制.实验研究已经发现[10],Nogo - B 的表达与LPA浓度密切相关,并且部分Nogo-B 通过LPA 介导的YAP 信号转导促进肿瘤发生.在这里,我们发现部分Nogo-B 通过与ATG5 合成NA,在很大程度上激活YAP 信号促进肿瘤进展.图3b 也显示了NA 浓度随时间演化出现了最大值,这是由于CEBPβ 增强了Nogo-B 的转录激活.
图3 [Nogo-B]与[NA]随时间演化的动力学关系Fig.3 Temporal evolutions of the levels of[Nogo - B]and[NA].
图4 呈现了[CEBPβ]随时间演化的动力学关系,以及[Nogo - B]随[CEBPβ]改变的函数关系.从图4a 可以看出,CEBPβ 极快地升高到了最大值,然后持续极短时间后急剧降低并趋于稳定.由此表明,CEBPβ 随时间演化的初始阶段呈现了脉冲式触发信号,增强了Nogo -B 的转录激活,提升了Nogo - B 表达水平.这种触发增强,是oxLDL 调节Nogo-B 激活的补充,CEBPβ通过短暂的触发信号促进Nogo -B 表达上调,与oxLDL 协同增强了oxLDL -Nogo -B -YAP 通路的激活.实验研究发现[7],CEBPβ 的表达提升的同时也增强了Nogo-B 的表达,Nogo -B 的表达水平与CEBPβ 的表达呈正相关.如图4b 所示,[Nogo - B]随[CEBPβ]的增加始于近似的线性增长,表明了Nogo -B 表达与CEBPβ 表达的正相关性,以及高表达的Nogo - B 只需较短时间CEBPβ 的刺激.从图4b 还可以看出,当CEBPβ浓度开始减少时,Nogo -B 呈现了趋于不变的稳态,并且稳态范围较大,直至浓度从38 nM 减少到15 nM 以内,Nogo-B 几乎趋于不变.这进一步表明了,CEBPβ 通过触发短暂的激活信号,与oxLDL 协同激活了稳定的oxLDL-Nogo -B -YAP通路[10].由此可以推断,CEBPβ 的敲减会在一定程度上阻碍oxLDL 诱导的Nogo-B 激活表达,从而影响YAP、CTGF 的激活表达水平.为了进一步确定YAP、CTGF 的激活表达对Nogo -B 的依赖性,可以考察[YAP]与[CTGF]随[Nogo -B]的变化关系.

图4 [CEBPβ]随时间演化的动力学关系( a) ,以及[Nogo - B]随[CEBPβ]变化的函数关系( b).Fig.4 Temporal evolution of the level of [CEBPβ]( a) ,and [Nogo - B]as a function of[CEBPβ]( b).
图5 显示了[YAP]与[CTGF]随[Nogo -B]变化的函数关系,如图5 所示,[YAP]和[CTGF]随[Nogo - B]的增大而增加表明,Nogo-B 的激活表达促使了YAP 和CTGF 的表达上调.由此可以确定,YAP 和CTGF 的活性依赖于Nogo-B.Nogo -B 诱导oxLDL 降解提升了YAP表达水平,并激活下游CTGF 等基因表达.从图5 还可以看出,当Nogo -B 浓度增加到一定值,然后当Nogo-B 浓度再减小时,YAP 和CTGF 都呈现了趋于不变的稳态,由此进一步表明,Nogo-B 表达上调诱导的oxLDL 降解,激活了稳定的oxLDL-Nogo -B -YAP -CTGF 通路.因此可以推断,Nogo-B 高表达所调控的信号通路途径中,Hippo 信号传导在异位Nogo -B 激活表达后被高度激活,这与Tian 等人[10]的实验结果相符.Nogo-B 通过促进oxLDL 降解,提升下游LPC 表达水平,由此导致LPA 在Nogo-B 表达上调后会显著增加,LPA 已被证明可以调节Hippo 途径信号[10,22].由于Nogo -B 通过提高LPA 表达水平来刺激YAP 活性,降低了YAP 的磷酸化,并增加了下游靶结缔组织生长因子( 例如CTGF 等) 的转录水平,因此,形成了基于Nogo -B 的稳定的信号通路.
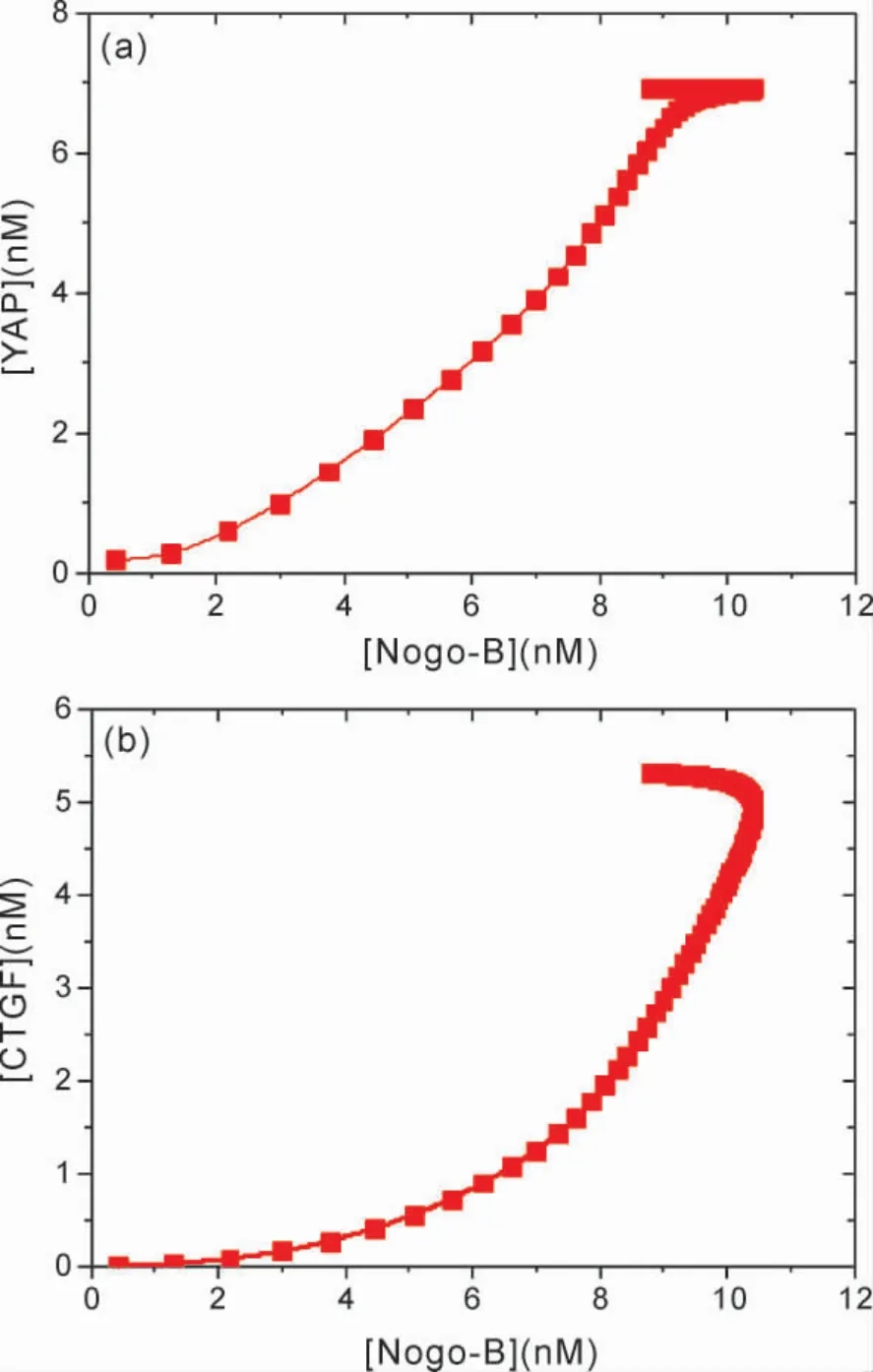
图5 [YAP]与[CTGF]随[Nogo -B]变化的函数关系.Fig.5 [YAP]and[CTGF]as a function of[Nogo-B]
实验研究[11,22]也表明JCAD 与Hippo 信号通路中关键分子LATS2 激酶相互作用,抑制YAP磷酸化.去磷酸化后YAP 进入细胞核,与TEAD分子结合,激活下游细胞增殖及肿瘤形成的相关因子( CTGF 等)[23,24].
图6 呈现了在不同 JCAD 浓度条件下,[CTGF]随时间演化的动力学关系.从图6 可以看出,[CTGF]随着时间演化,逐渐升到了较大的稳定值,表明CTGF 被激活并达到一定的表达水平.比较图中不同JCAD 浓度时的[CTGF]幅值,可以发现,[JCAD]=15 nM 时的[CTGF]幅值明显高于[JCAD]为5 nM 时的幅值,这表明,在较高的JCAD 浓度条件下对CTGF 的激活幅度,大于在低浓度下对CTGF 的激活幅度,并且,随着JCAD 浓度的增加,CTGF 增加到稳定值的时间变短.由此表明,JCAD 在较大程度上调控着CTGF 的激活速度与表达水平.这是由于JCAD 与LATS2 相互作用,增强了在Hippo 途径中YAP 去磷酸化的能力,进而上调了CTGF 等HCC 细胞增殖因子.然而,沉默的JCAD 会产生相反的效果,JCAD 的低水平表达则会降低与LATS2 的结合作用,使得LATS2 促使YAP 磷酸化的能力增强,抑制HCC 的生长增殖.实验研究发现[11,17],在正常肝组织中,JCAD 一直维持低表达水平,在NASH-HCC 标本中JCAD 的表达水平明显偏高.因此,JCAD 的过表达促进了肿瘤的生长和增殖,表明了JCAD 在NASH 向HCC 过渡期间,激活Hippo 信号级联反应中的新作用.

图6 不同[JCAD]条件下,[CTGF]随时间演化的动力学关系.Fig.6 Temporal evolutions of the level of[CTGF]at different[JCAD].
JCAD 通过与激酶LATS2 结合反应,调控YAP 的磷酸化.在Hippo 信号通路中,LATS2 是调控YAP 的磷酸化水平的另一个重要调节子,其功能与JCAD 相反[11].为了进一步研究激酶LATS2 在Hippo 信号通路中抑制NASH 发展为HCC 中的作用,可以考察不同FLATS2 浓度条件下[CTGF]随时间的演化关系.
图7 显示了在不同FLATS2 浓度条件下,[CTGF]随时间演化的动力学关系.比较图中FLATS2 浓度在[FLATS2]=5 nM、[FLATS2]=10 nM 和[FLATS2]=15 nM 时的CTGF 浓度幅值,可以发现,[FLATS2]=15 nM 时的CTGF 幅值明显低于浓度为[FLATS2]=5 nM 时的幅值,这是由于随着FLATS2 的增加,FLATS2 对CTGF的抑制程度增加.与JCAD 调控效应相反,随着FLATS2 浓度的减少,CTGF 增加到稳定值的时间变短,浓度幅值变大.由此表明,较高浓度的FLATS2 则会较大程度地促进YAP 磷酸化,抑制YAP 活性,进而降低了CTGF 等增殖因子的表达水平,较为显著地抑制了HCC 细胞的生长增殖.因此可以得出,LATS2 确实可以充当Hippo 信号通路的下游调节因子,从而影响HCC 细胞的增殖.Ye 等人[11]的实验的确发现,在Hippo 信号通路中,LATS2 可以使YAP 磷酸化以抑制CTGF 等表达,进而抑制脂肪肝病诱导的HCC 发生发展.
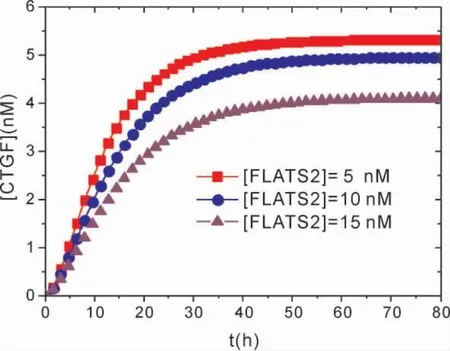
图7 不同[FLATS2]条件下,[CTGF]随时间演化的动力学关系.Fig.7 Temporal evolutions of the level of[CTGF]at different[FLATS2].
4 结 语
本文建立理论模型研究Nogo -B 与JCAD 诱导非酒精HCC 基因激活.理论模型考虑: ( 1)Nogo-B 通过激活DoxLDL - Nogo - B - YAP 通路[12],促使HCC 发生发展; (2) JCAD 与LATS2相互作用抑制YAP 磷酸化,未被磷酸化的YAP激活Hippo 信号通路中下游与肿瘤形成相关的增殖因子,导致HCC 的发生发展.研究发现,ox-LDL 在很短的时间内降解,产生了大量降解后的DoxLDL,进而很快激活下游通路信号( LPC、ATX、LPA 等) ,在很大程度上提升了Hippo 信号通路中YAP 的活性,激活下游CTGF 等癌基因.通过分析Nogo-B 随时间演化的动力学特性,可以发现,Nogo - B 与ATG5 的结合反应放大了Nogo-B 表达信号,促进LPA 合成并激活YAP.作为oxLDL 激活Nogo -B 的补充,CEBPβ 呈现了脉冲式触发信号增强了Nogo -B 的激活表达,并促进oxLDL 迅速降解,协同增强了稳定的Dox-LDL-Nogo-B -YAP -CTGF 通路信号激活.另外,JCAD 蛋白可以与LATS2 相互作用,促使YAP 去磷酸化,去磷酸化的YAP 上调CTGF 等增殖因子,表明了较高浓度的JCAD 调控CTGF 表达水平提升.与JCAD 调控效应相反,FLATS2 通过抑制YAP 活性,降低CTGF 等增殖因子的表达水平.由此也表明了JCAD、LATS2 在NASH 向转变HCC 过程中,激活Hippo 信号通路的新作用.事实上,Nogo -B 可以诱导oxLDL 的自噬和降解[25],在NASH-HCC 的转化过程中,脂肪变性发展和自噬功能降低均与NASH 有关[26,27],炎症和内质网应激也会在很大程度上影响NASH 的发病[28],Nogo-B 的缺失会减少原发性HCC 中STAT3 的磷酸化,这样Nogo -B 还可以通过调节IL-6/STAT3 信号影响HCC 的发生发展.病理研究数据表明,未折叠的蛋白质反应UPR( unfolded protein response) 激活,在促进非酒精性脂肪肝病发展为HCC 进程中也会起着很重要的作用[29-31].本文中只考虑Nogo -B 与JCAD 诱导HCC 基因激活的一种致癌机制,理论结果符合实验[10,11],表明了Nogo-B 表达上调,会在很大程度上促使HCC 发生发展,以及JCAD 蛋白与LATS2 激酶也在很大程度上调控CTGF 等HCC 细胞的增殖,进而调节癌基因表达通路的动力学行为.
