miR-30家族在心肌缺血再灌注损伤中作用的研究进展
2022-03-04刘思娜仇盛蕾刘鑫毅周莹洁杨悦文尚菊菊
刘思娜,仇盛蕾,刘鑫毅,周莹洁,杨悦文,尚菊菊
心肌缺血再灌注损伤(myocardial ischemic reperfusion injury,MIRI)是指心肌持续缺血后,恢复血液灌注,虽然使大部分濒临坏死的心肌得以恢复,有效限制了心肌梗死范围及心室重构,但也会导致心肌顿抑、室性心律失常、微血管功能障碍甚至心肌细胞死亡、心力衰竭等不良事件的发生,造成部分心肌损伤进行性加重的现象[1-3]。研究表明,最终心肌梗死面积中有高达50%的部分与MIRI的发生有关[4]。MIRI严重影响了急性心肌梗死(acute myocardial infarction,AMI)病人再灌注治疗的临床效果和预后,寻求防治MIRI的有效措施已成为目前研究的重要目标之一。
目前,MIRI的发生机制尚未完全阐明,研究证实其与氧化应激、钙超载、自噬、细胞凋亡和坏死等密切相关[5]。随着对疾病认识的深入及技术的发展,针对疾病治疗的研究现已扩展到微观分子水平。微小RNA(microRNA,miRNA)作为表观遗传学的一部分已成为新的研究热点[6]。研究发现,多种miRNA具有心肌保护作用,其中microRNA-30(miR-30)家族是心肌中表达最丰富的miRNA物种之一,其表达水平与MIRI密切相关[7]。现就有关miR-30家族在MIRI中作用的研究进展进行综述。
1 miR-30家族概述
miR-30家族是miRNA家族中的一个重要成员,由miR-30a、miR-30b、miR-30c、miR-30d和miR-30e组成,miR-30c又分为miR-30c-1和miR-30c-2,由位于人类染色体1、6和8的6个基因编码,并在5′端共享一个种子序列,但在3′端有不同的补偿序列,这使得miR-30家族成员可以针对不同的基因和途径执行不同的生物学功能[8-9]。一系列研究表明,miR-30家族在MIRI的自噬、凋亡、坏死等病理过程中发挥不同程度的调节作用[10-14]。研究显示,miR-30家族在AMI病人或小鼠梗死心肌及心肌细胞缺氧模型中均表达上调[15-17];而在MIRI病人或小鼠缺血再灌注(ischemia/reperfusion,I/R)心肌及心肌细胞缺氧/复氧(hypoxia/reoxygenation,H/R)模型中表达下调[10,18]。当miR-30家族表达水平失调时,可通过调节不同的靶基因来介导相应的机制;相应的,通过调控miR-30家族的水平来调节基因的表达,可达到减少MIRI的目标。miR-30家族在MIRI治疗中的应用日益成为研究热点。
2 miR-30家族参与MIRI的机制研究
2.1 miR-30家族调节心肌细胞自噬 既往研究发现,自噬参与了MIRI的病理过程。研究表明,在早期心肌缺血阶段,自噬对于缺血心肌具有一定的保护作用,帮助细胞度过能量危机;而再灌注期间由于自噬相关蛋白Beclin-1的过度表达,自噬被过度激活,进而导致必需的细胞成分过度降解,细胞损伤加重,从而促进了心肌细胞坏死的发生[19-20]。
关于miRNAs对自噬的调控作用的首次报道是在2009年,Zhu等[21]发现,在肿瘤细胞中自噬特异基因Beclin-1是miR-30a的潜在靶点,miR-30a可通过与Beclin-1的信使RNA3′端的序列(3′untranslated regions,3′UTR)结合,抑制Beclin-1基因表达,进而削弱自噬水平。曾召林等[22]研究报道miR-30家族参与了自噬的诱导激活、囊泡成核、囊泡延伸的不同阶段。晏浩等[14]研究发现:在H/R心肌细胞中miR-30a表达水平明显下降,Beclin-1蛋白及自噬标志蛋白-微管相关蛋白1轻链3-β(microtubule-associated protein 1 light chain 3-β,MAP1 LC3-Ⅱ,LC3-Ⅱ)与微管相关蛋白1轻链3-α(microtubule-associated protein1 light chain3-α,MAP1 LC3-Ⅰ,LC3-Ⅰ)的比值即LC3-Ⅱ/LC3-Ⅰ比值增加;同时过表达miR-30a可显著抑制Beclin-1蛋白表达及LC3-Ⅰ向LC3-Ⅱ的转化,进而减少自噬来减少心肌细胞死亡;过表达miR-30a还可减少Beclin-1 的C端片段Beclin-1C形成而抑制凋亡。此外,研究还阐述了在MIRI中自噬和凋亡两种类型的细胞程序性死亡之间拮抗和协同关系的机制:①凋亡相关蛋白半胱氨酸蛋白酶(Caspase)介导Beclin-1裂解形成的Beclin-1C,可抑制Beclin-1诱导的自噬,并通过促进线粒体释放促凋亡因子来促进细胞凋亡[23];②MIRI时激活的c-Jun氨基末端激酶(c-Jun NH2-terminal kinase,JNK)可促进Beclin-1与抗凋亡蛋白Bcl-2结合体的释放而增加自噬[24]。
Yang等[25]发现:①在AMI病人血清和缺氧心肌细胞中,miR-30a高度富集,且可通过外泌体在心肌细胞间有效转运;②抑制缺氧诱导因子-1α(hypoxia-inducible factor 1 alpha subunit,HIF-1α)抑制miR-30a上调;③抑制miR-30a或外泌体释放可增加Beclin-1、自噬相关蛋白12(autophagy-related protein 12,Atg12)的含量和LC3Ⅱ/LC3Ⅰ比值,从而维持心肌细胞缺氧后的自噬反应;④抑制miR-30a或外泌体释放可减轻缺氧诱导的心肌细胞凋亡,但其具体机制有待进一步研究。此研究虽在缺氧细胞中进行,但表明了外泌体在miR-30a介导的自噬中的作用。
王竟靖等[26-27]研究发现:①相对于正常组,在缺氧条件下,miR-30a、Beclin-1和LC3-Ⅱ表达均上调(P<0.05);与正常组和缺氧组比较,在复氧时,miR-30a表达下调,Beclin-1和LC3-Ⅱ表达上调(P<0.05),表明心肌细胞在缺氧条件下发生自噬,而在H/R过程中自噬进一步增强;②进一步研究发现,长链非编码RNA(long noncoding RNA,lncRNA)AK088388在H/R过程中表达上调,并可通过竞争性结合miR-30a,促进Beclin-1和LC3的表达,进而增强再灌注过程中自噬的发生,最终导致心肌细胞损伤。此研究表明了lncRNA在miR-30a介导的MIRI自噬中的作用。综上所述,miR-30家族在MIRI自噬中的调节作用主要与miR-30a有关。在缺血再灌注阶段,miR-30a的下调可导致Beclin-1表达的提高而引起过度自噬,从而加重细胞损伤;而过表达的miR-30a可下调Beclin-1而抑制自噬泡成核过程,进而减轻心肌细胞过度自噬,改善MIRI。此外,lncRNA AK088388可通过下调miR-30a而增强自噬进而加重MIRI。
在AMI病人血清和缺氧心肌细胞中,miR-30a、Beclin-1表达及自噬水平均上调;抑制miR-30a上调或外泌体释放可导致Beclin-1表达水平的升高而进一步维持自噬反应。但如果进一步推测,缺氧条件下上调的miR-30a是否不影响Beclin-1表达及自噬水平,或者由于此阶段的自噬主要与AMP活化蛋白激酶(AMP-activated protein kinase,AMPK)有关[19]。故上调的miR-30a虽可导致Beclin-1表达下调,但下调程度不足以影响自噬,有待进一步研究。
2.2 miR-30家族调节心肌细胞凋亡和坏死 研究发现,缺血再灌注期间同时存在坏死和凋亡[28]。在缺血早期,凋亡是细胞死亡的主要形式,而在缺血晚期,大部分心肌细胞死亡是由缺血坏死引起的。miR-30家族不仅调控自噬,还参与细胞凋亡和坏死。
Li等[29]研究发现:①氧化应激(oxidative stress)时,miR-30家族的表达水平降低,而肿瘤抑制因子p53表达上调,p53可转录激活线粒体分裂蛋白——动力蛋白相关蛋白-1(dynamin-related protein 1,Drp1),后者可引起线粒体外膜产生的碎裂片增多,并通过启动线粒体分裂程序传递p53凋亡信号;②p53是miR-30家族的潜在靶点,miR-30家族可通过与野生型p53的3′UTR结合,抑制p53及其下游靶基因Drp1的表达来抑制线粒体分裂,进而抑制细胞凋亡。此研究虽然不是在MIRI模型中展开,但却首先报道了miR-30家族影响心肌细胞凋亡的分子机制。
Forini等[13]研究发现:①在I/R大鼠心肌细胞中,三碘甲状腺原氨酸(triiodothyronine,T3)水平降低,而T3给药可抑制三磷酸腺苷(adenosine triphosphate,ATP)生成率和细胞色素C氧化酶(cytochrome-C oxidase,COX)活性的下降以及线粒体超氧化物歧化酶的升高,即T3可挽救线粒体的活性并限制超氧化物的形成,从而发挥抗线粒体氧化损伤的作用;②在I/R早期T3水平降低的同时,伴随着miR-30a下调以及其靶点p53的上调,而p53可通过激活凋亡调节蛋白——Bcl-2相关蛋白(bcl-2-associated X protein,Bax)促进线粒体介导的细胞死亡,包括通过促进线粒体外膜通透化(mitochondrial outer membrane permeablisation,MOMP),进而促进细胞色素C(cytochromes C,Cyt-C)的释放引起的细胞凋亡,以及通过驱动线粒体膜通透性转换孔(mitochondrial permeability transition pore,mPTP)导致线粒体内膜跨膜电位(mitochondrial transmembrane potential,delta psi M,△Ψm)丧失,ATP耗竭而引起的细胞坏死,此外,p53可直接增强缺血后线粒体的损伤,从而驱动mPTP和细胞色素C的释放而引起细胞死亡;③采用T3处理后可逆转上述现象,而miR-30a基因敲除后可明显抑制T3的调节作用,即T3可通过诱导miR-30a上调,抑制p53的激活,进而抑制Bax的过度表达,使得线粒体膜去极化受限,线粒体功能得以保留,凋亡和坏死程度降低,从而限制I/R损伤。总之,此研究提示一种由T3介导的以线粒体为靶点的通过调节miR30a/p53轴发挥心脏保护作用的新机制。
Wang等[12]研究表明,在过氧化氢处理后的心肌细胞和小鼠I/R心肌中,亲环素D(cyclophilin D,CypD)水平升高,其基因的敲除可抑制心肌细胞坏死,并减少心肌梗死,保护缺血再灌注损伤的心功能;miR-30b水平降低,miR-30b过表达可降低CypD的表达,从而减少CypD介导的I/R心肌细胞坏死数量;转录因子E2F1水平升高,其可通过抑制miR-30b表达,上调CypD水平,进而促进心肌细胞坏死,而这些作用可被E2F1的下调所拮抗,即E2F1的敲除可通过在转录水平负调控miR-30b来抑制心肌坏死。总之,此研究结果揭示了一个由E2F1、miR-30b和CypD组成的新的心肌细胞程序性坏死调节通路。
因为CypD是mPTP的必需成分之一,故结合以上3项研究可以得出,miR-30家族可以通过调节线粒体形态和影响线粒体功能达到对MIRI的调控作用[12-13,29-31]。Kim等[32]研究发现:①在心肌梗死小鼠模型中,miR-30-5p家族的表达显著下调(此结论与先前在缺氧时miR-30家族的表达上调的结论相反,有待进一步研究);②单个miR-30-5p家族成员在缺氧条件下具有抗凋亡作用,并且直接靶标促凋亡基因Picalm和Skil;③Picalm和Skil的敲除显著上调磷酸化蛋白激酶B(p-Akt)和Bcl-2的表达,而显著下调Caspase-3的表达。已有研究报道Skil可以直接结合并激活p53来诱导凋亡[33]。故此研究表明,过表达miR-30-5p家族可通过直接靶向抑制Picalm和Skil的表达来提高p-AKT和Bcl-2的表达水平,进而抑制Caspase-3的表达而减少心肌细胞凋亡;或者通过抑制Skil直接下调p53的表达,进而抑制心肌细胞凋亡,从而保护心肌细胞。虽然此研究并未在I/R模型中进行,但发现了miR-30-5p家族调控凋亡的新通路,提示研究时需要注意考虑miR-30家族亚型的独特调节。
过表达的miR-30家族可通过减少其靶向因子p53、CypD、Picalm、Skil等的表达,进而抑制MIRI的细胞凋亡和坏死而保护心肌细胞。T3可以上调miR-30家族的表达,E2F1可以下调miR-30家族的表达。
2.3 miR-30家族与其他 Gambacciani等[18]研究发现,在活体大鼠心肌梗死和I/R模型中,miR30家族成员的水平下调,而DNA甲基转移酶3a(DNA methyltransferase 3a,DNMT3a)的蛋白水平升高;同时过表达的miR-30c可通过直接与DNMT3a的3′UTR相互作用而导致DNMT3a酶在mRNA和蛋白水平上降低。此研究结果提示miR-30家族可能通过调控DNMT3a,进而调控DNA甲基化的改变而改善MIRI的新机制,其涉及缺血性心肌损伤后的表观遗传学调控。DNA甲基化与微小RNA同属于表观遗传学的一部分,受DNMT的调控,MIRI时大量基因的甲基化水平发生变化,导致基因的表达异常,加剧心肌损伤,而DNA甲基化抑制剂能有效改善MIRI,虽然目前在临床上还没有甲基化抑制剂应用于心脏疾病治疗的研究,但基于miR-30c的可改善MIRI的甲基化抑制剂的研究值得期待[34]。
Zheng等[10]研究发现,在MIRI病人的外周血中miR-30e表达降低;沉默miR-30e (si-miR-30a)的作用机制:①抑制H9c2细胞凋亡,降低凋亡调节因子Bax蛋白表达、Caspase-3活性;②显著增加自噬相关蛋白LC3、p62、Beclin-1的表达;③增加Notch受体Notch1及其靶基因——碱性-螺旋-环-螺旋(basic helix-loop-helix,bHLH)转录抑制因子Hes1和p-Akt的表达;④降低诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)的表达,增加氧化应激相关蛋白如超氧化物歧化酶(superoxide dismutase,SOD)、谷胱甘肽(glutathione,GSH)、谷胱甘肽过氧化物酶(glutathione peroxidase,GSH-Px)的活性;用自噬抑制剂3-甲基腺嘌呤(3-methyladenine,3-MA)抑制自噬后,可逆转si-miR-30e的①②④作用;促进Notch1表达后,可显著增强si-miR-30e的①③④作用。miRNA-30e通过自噬和Notch1/Hes1/Akt信号通路在抑制细胞凋亡和氧化应激中,保护心肌免受I/R损伤。
3 中医药调控miR-30家族防治MIRI研究
2016年,Li等[35]研究发现,在小鼠I/R心肌细胞中,miR-30a的表达明显下调,Beclin-1、LC3-Ⅱ的表达上调;丹酚酸B(salvianolic acid B,Sal B)可通过上调内源性miR-30a的表达,降低Beclin-1、LC3-Ⅱ的表达及乳酸脱氢酶(lactate dehydrogenases,LDH)漏出率,并提高p-Akt的表达及心肌细胞活力,从而减轻心肌自噬,保护心肌细胞;抑制磷脂酰三磷酸肌醇(phosphatidylinositol 3-Kinases,PI3K)表达(PI3K抑制剂LY294002处理后)可使Beclin-1的表达增加,p-Akt蛋白表达减少。提示Sal B诱导的miR-30a抗自噬作用机制可能与PI3K/Akt信号通路有关。此研究表明了中药有效成分Sal B在miR-30a介导的MIRI自噬中的作用。
Liu等[36]研究发现,在大鼠I/R心肌模型和H/R心肌细胞模型中,miR-30a表达增高,Beclin-1的表达水平和LC3BⅡ/LC3BⅠ比值降低,自噬受到抑制,心肌细胞凋亡增加;蛇床子素(osthole)可减少心肌细胞胶原含量,减轻心肌肿胀、坏死和心肌纤维化,从而减缓心肌损伤;蛇床子素可通过下调miR-30a的表达,提高Beclin-1的表达水平而增强自噬,并可减轻心肌细胞凋亡;自噬抑制剂3-MA显著逆转蛇床子素对细胞凋亡的保护作用,提示自噬在蛇床子素抑制H/R细胞损伤中起重要作用。此研究表明了蛇床子素在miR-30a介导的MIRI自噬中的作用,但该研究结果与上述研究有争议,有待进一步验证。
Wang等[37]研究发现:①在叔丁基过氧化氢(tert-Butyl hydroperoxide,TBHP)诱导的H9c2大鼠I/R心肌细胞中,miR-30c-5p的表达水平下调;②三七总皂苷(panax notoginseng saponins,PNS)及其活性成分人参皂苷Re可通过显著上调miR-30c-5p的表达水平,抑制p53的表达而减少细胞凋亡,并可增加心肌细胞存活率及减少LDH释放而减轻细胞损伤,从而改善MIRI。Sal B和PNS可通过上调miR-30家族的表达分别减轻心肌细胞自噬与凋亡,而蛇床子素通过下调miR-30a的表达增强自噬而减轻心肌细胞凋亡,进而改善MIRI。详见图1。
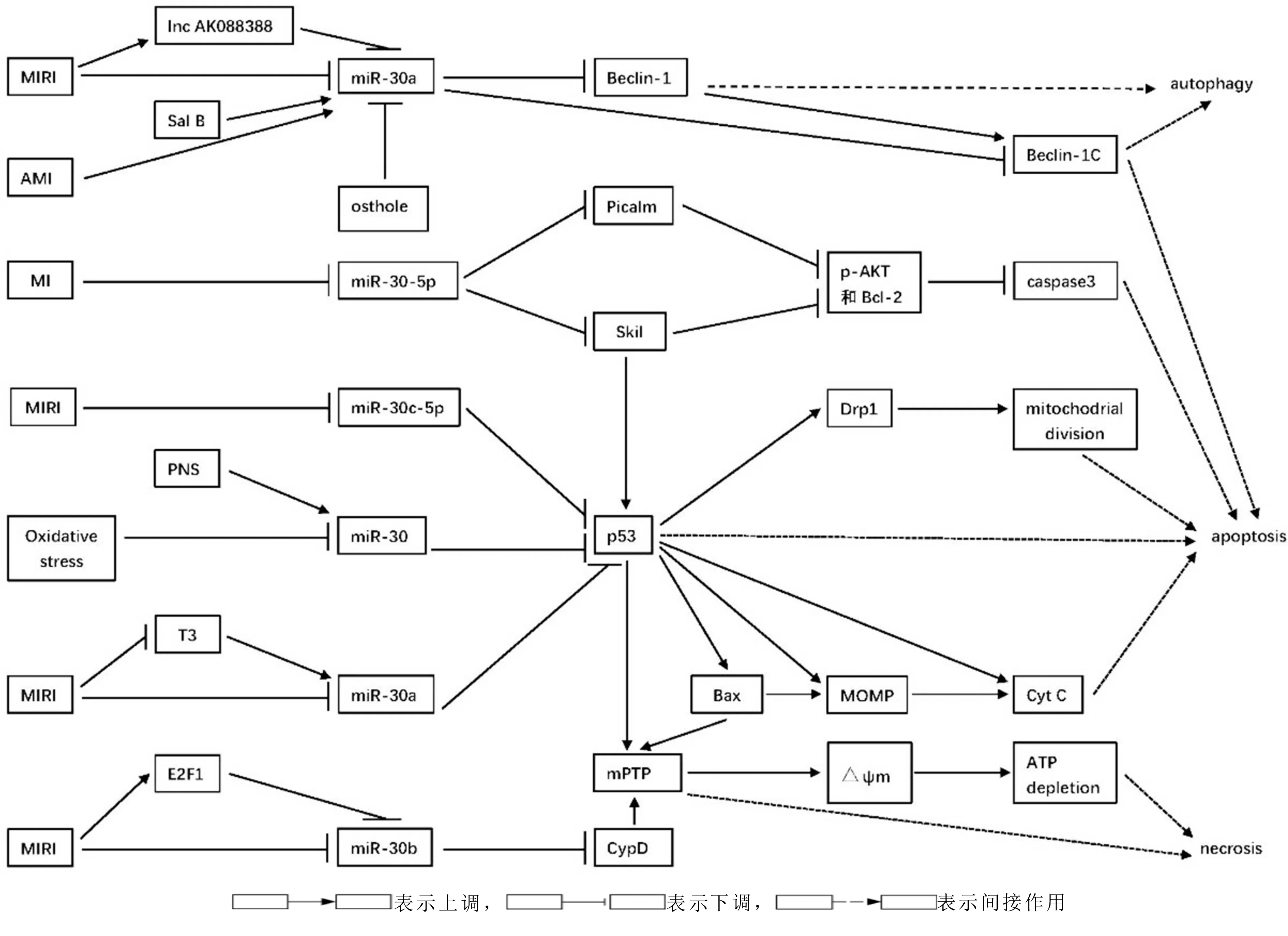
图1 miR-30家族在心肌缺血再灌注损伤中的研究
4 小 结
miR-30家族作为非编码RNA中的一员与心肌I/R损伤的关系密切。miR-30家族通过调节相关基因及其调节因子表达而参与影响MIRI的自噬、凋亡、坏死等病理过程。总体而言,在发生MIRI时miR-30家族表达下调,而通过上调miR-30家族的表达可改善MIRI。此类基于表观遗传学调控的病理生理学相互作用的分子和细胞效应的研究,将为不断完善MIRI病理机制,研究新的诊断标志物和治疗靶点,进一步开发抗MIRI新药提供实验依据和理论依据。
中医药治疗作为我国的特色医疗手段,已经成为MIRI治疗中不可缺少的组成部分[38]。丹酚酸B、蛇床子素、三七总皂苷等中药有效成分可通过调节miR-30的表达水平进而影响MIRI,此类强调基因和环境因素等相互作用的表观遗传学调控的研究,对解释及丰富中医“证候”“天人相应”的发病观以及“辨证论治”的个体化治疗观等中医理论具有重要意义;对阐明中医药治疗MIRI的分子水平作用机制,即单药作用多靶点效应及中药配伍、复方等多药联合调配后的发挥整体药效的多靶标共同作用提供了新的切入点;对创新中药新药研发以及研究中医药治疗心血管疾病及其相关预后干预带来新思路[6,39-40]。
然而,目前关于miR-30家族在MIRI中的作用研究尚处于初始阶段,且miR-30家族在MIRI中的作用关系大都集中在体外研究,序列的不保守性和一些实验方法的限制阻碍了体内实验的进展,大部分研究还局限在基础实验阶段,其临床效果还有待进一步验证。未来需要更多的研究去探索揭示MIRI的机制,并需要将这些研究成果与临床实际相结合,为MIRI的防治提供新思路与新策略。
