呋喃[3,2-b]吲哚类化合物碱性条件下扩环重排反应
2022-03-04张志国曹夕阳王港方世亮吴昊张贵生
张志国,曹夕阳,王港,方世亮,吴昊,张贵生
(河南师范大学 化学化工学院,河南 新乡 453007)
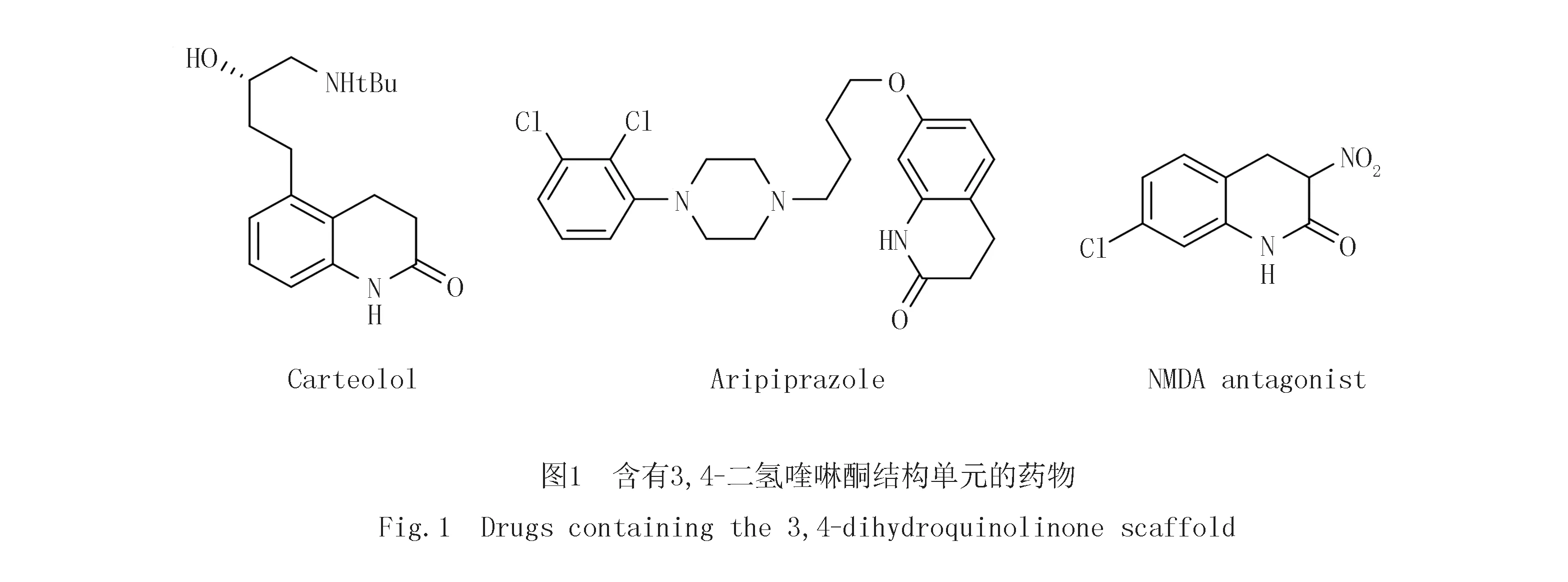
2-喹啉酮是一类非常重要的含氮六元杂环结构单元,广泛存在于天然产物、功能有机材料、生物活性化合物和商业化学品中,作为核心结构调节这些化合物的各种结构性能(图1)[1].它们还是重要的有机合成中间体,用于构建各种含氮、氧等杂原子的功能有机杂环化合物[2-3].目前,人们利用传统合成方法,结合电化学[4-5]、光化学[6-7]等现代有机合成化学手段,通过Friedel-Crafts环化[8],无催化剂[5],过渡金属催化的串联反应和自由基反应[9-10]等反应发展了分子内和分子间环合反应,多组分反应等多种方法构建了2-喹啉酮结构单元.
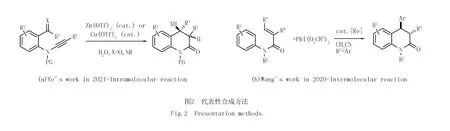
最近,文献[11]利用铜催化或者锌催化的邻位带有亚胺和炔胺侧链的芳基化合物与水或者胺类的炔烃水合/分子内曼尼希串联反应,高选择性地合成了3,4-二氢-2-喹诺酮类化合物.该转化过程是利用分子内环合反应策略制备2-喹啉酮类化合物的典型案例[12-15](图2(a)).2020年,文献[16]发展了铼催化的N-芳基丙烯酰胺类化合物与三价高价碘试剂的烷基芳基化反应.他们在加热条件下通过串联自由基加成/环化反应实现了3,4-二氢喹啉酮类化合物的合成.这一转化具有广泛的底物范围及良好的官能团耐受性,可以认为是过渡金属促进的分子间串联环合反应制备2-喹啉酮类化合物的典型代表[17-21](图2(b)).虽然2-喹啉酮类化合物的构建取得了显著进展,但是现有方法通常存在反应条件苛刻、底物适用范围有限和产物产率低等缺点.因此,发展2-喹啉酮类化合物简单、高效的合成方法,同时兼顾绿色化学、原子经济性化学和可持续性化学仍然是一项具有挑战性的工作.
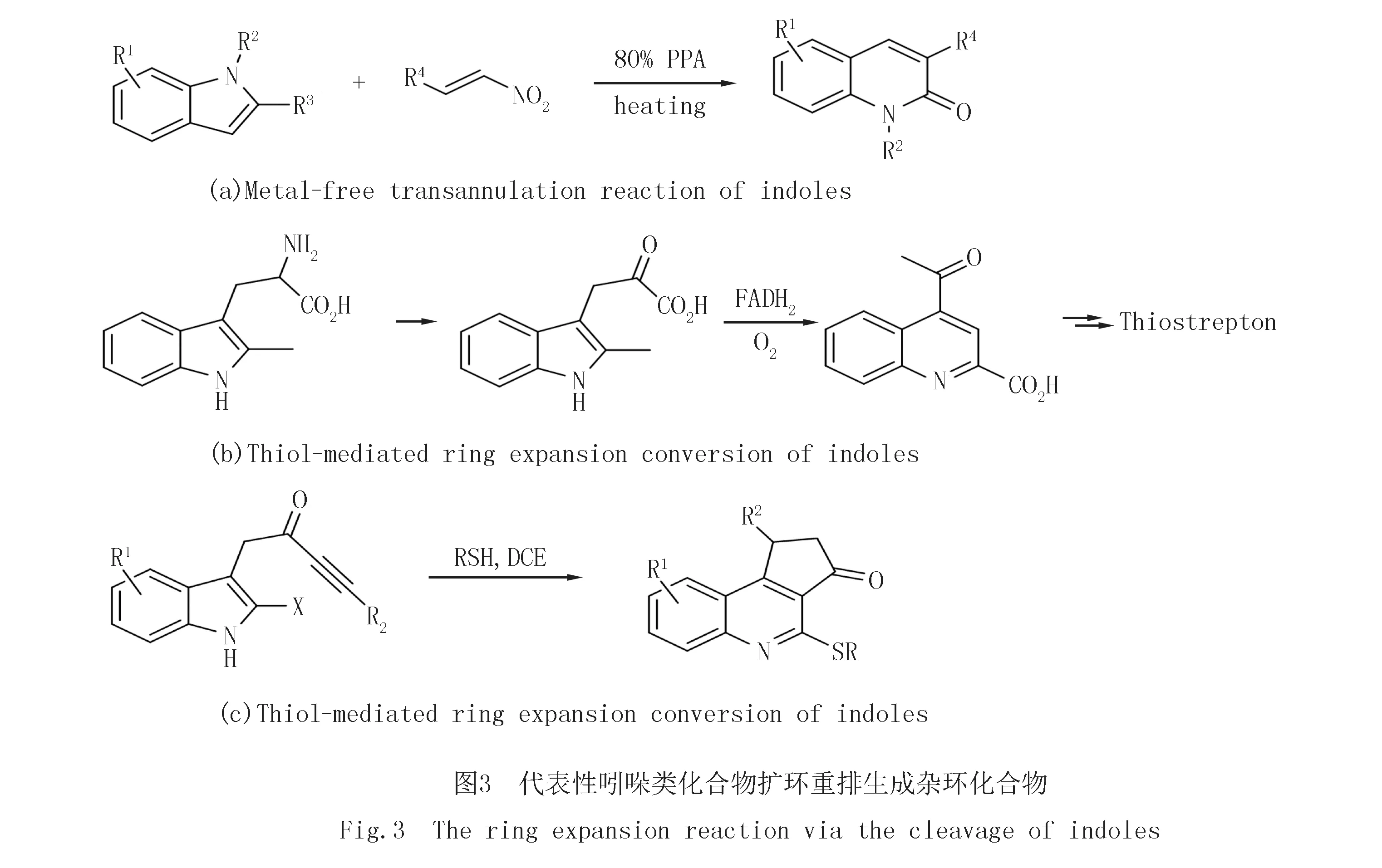
吲哚是一类非常重要的具有苯并五元氮杂环结构单元的有机化合物,它们广泛存在于天然产物、医药、农药、香料、染料和功能材料中.同时,它们也是获取其他精细化学化工产品的重要有机合成中间体.吲哚类化合物可以发生扩环重排反应生成多种含氮、氧等杂原子的苯并六元、七元甚至八元环的化合物[22-25], 其中包括一类非常重要的生成喹啉类化合物的扩环反应[26-35].Alexander课题组[29,33]陆续报道了吲哚与硝基烯烃在无金属条件下合成2-喹喏酮类化合物的分子间反应(图3(a)).Thiostrepton 是一种抗生素,Quinaldic acid是它的重要活性结构单元.文献[26]从2-甲基色氨酸出发,巧妙地利用侧链与吲哚部分的氧化扩环反应获得了可转化为Quinaldic acid结构单元的化合物4-乙酰基喹啉-2-酸(图3(b)).最近,文献[27]描述了一种条件温和、操作简单和高转化率的三步串联方法,可以将吲哚炔酮直接转化为功能化的喹啉衍生物.其中,单一的“多任务”硫醇试剂促进了脱芳构化螺环化、亲核取代和单原子扩环三步串联反应(图3(c)).总之,通过分子间或者分子内反应首先获得一个带有功能侧链的吲哚前体化合物,然后再通过侧链参与的扩环重排反应将吲哚转化成喹啉是这类合成策略的共同特点.
本课题组一直专注于含氮和氧等杂原子的杂环类化合物的合成研究工作[36-39],陆续报道了Ag(I)促进的羰基-N-丙炔酰胺衍生物通过分子内[3+2]环加成反应合成呋喃[3,2-b]杂环化合物的新策略[36-37].2012年,课题组从N-芳基炔丙胺衍生物出发,在氧化银和氢氧化钾的共同作用下,通过芳基侧链上的炔丙基全碳1,3-偶极子与羰基的[3+2]环加成反应获得了一系列呋喃[3,2-b]吲哚类化合物[36].基于文献报道的吲哚类化合物多样的生物活性,希望能够脱除呋喃[3,2-b]吲哚类化合物氮原子上酰基保护基,然后再用糖类化合物修饰氮原子,期望获得一类新的生物活性分子.结果,当尝试用氢氧化钠在水和乙醇等体积比的混合液中水解N-乙酰基呋喃[3,2-b]吲哚(1a)的酰胺结构单元时,没有得到呋喃[3,2-b]吲哚预期产物2a,而是得到了另一种白色固体化合物(图4(a)).单晶衍射结果表明反应生成了4 位带有亚甲羟基的喹啉酮类化合物 4-(羟甲基)-4-甲基-3,4-二氢喹啉-2(1H)-酮(3a,CCDC NO.2124765,相关晶体学数据见表1).值得一提的是,1972年WARNHOFF等人[40]发现在回流乙酸和盐酸混酸的条件下,可以促使侧链上带有羰基的吡咯酮并吲哚类化合物4发生一种新的分子内缩合/重排串联反应生成喹啉酮联吲哚类化合物5(图4(b)).WARNHOFF等人的研究结果激发了课题组继续开展研究的兴趣,因为一方面课题组所采用的底物和他们的底物比较相似,但产物不同.另一方面课题组的重排扩环反应是在碱性条件下发生的,而WARNHOFF等人[40]的反应是在酸性条件下发生的.
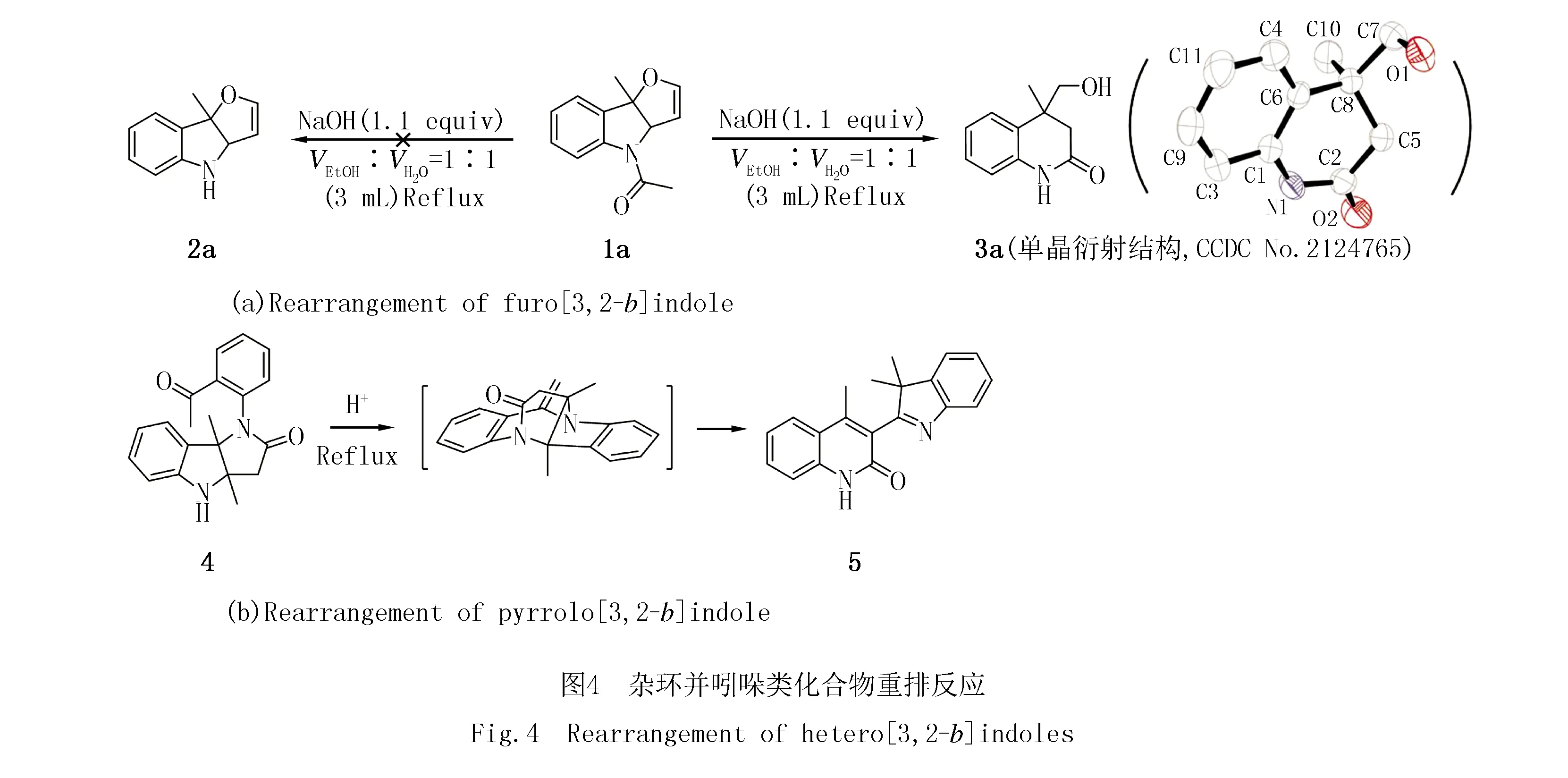
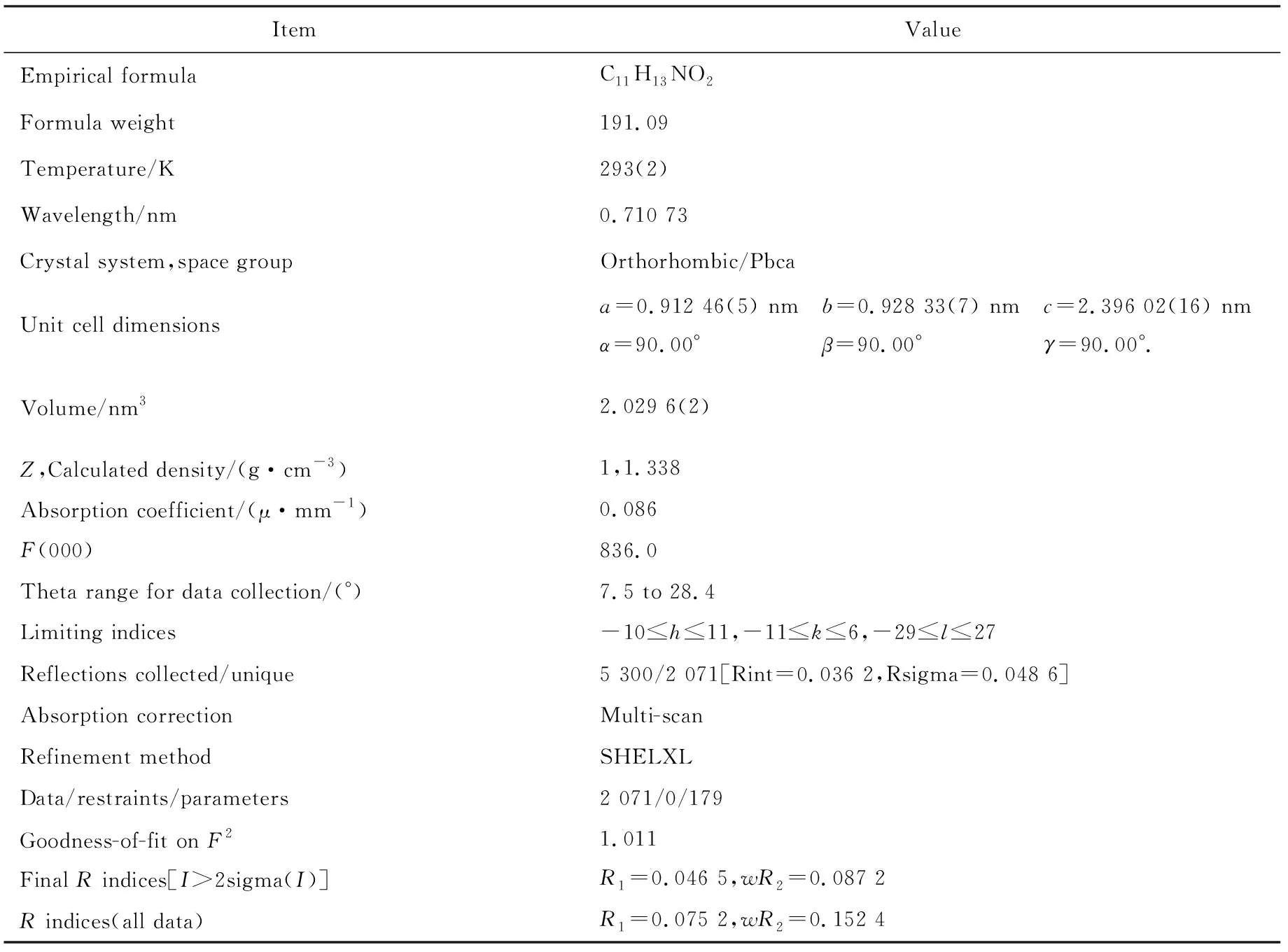
表1 化合物3a(CCDC No.2124765)的主要晶体学数据
1 实验部分
1.1 仪器与试剂
所有试剂均购自商业来源,使用前没有进一步纯化,除非另外说明.所有反应通过用GF254硅胶涂覆的TLC(薄层色谱)板监测.本文中所有化合物均是通过使用200~300目硅胶填充柱色谱法分离纯化.1H NMR和13C NMR光谱使用(Bruker Avance)400 MHz光谱仪测量,使用四甲基硅烷作为内标,DMSO-d6,CDCl3为溶剂,δ值以ppm为单位给出,偶合常数(J)以Hz为单位给出.高分辨率质谱在MicroTOF质谱仪上测得.化合物的熔点是在配备有温度计的熔点仪上测量并且温度未经校正.
1.2 实验方法
向配备有球形冷凝器(40 cm长)的圆底烧瓶(25 mL)中加入呋喃[3,2-b]吲哚1a(108 mg,0.50 mmol),KOH(59 mg,1.05 mmol)和乙二醇和水(V乙醇∶V水=1∶1)3 mL混合,然后将混合物在油浴中回流充分搅拌6 h,冷却后,向混合物中加入水(15 mL),用正丁醇萃取(每次4 mL,萃取3次).减压除去溶剂(油泵),残余物通过硅胶柱色谱纯化,得到化合物3a(68 mg,71%),为淡黄色固体(洗脱剂:V石油醚∶V乙酸乙酯∶V乙醇=10∶6∶1).
1.3 产物表征数据
4-(羟甲基)-4-甲基-3,4-二氢喹啉-2(1H)-酮(3a-3e):淡黄色固体,m.p.151~152 ℃;1H NMR(400 MHz,CDCl3)δ8.72(s,1H),7.28(d,J=7.6 Hz,1H),7.20(t,J=7.3 Hz,1H),7.06(t,J=7.4 Hz,1H),6.83(d,J=7.8 Hz,1H),3.70(dd,J=10.9,4.2 Hz,1H),3.53(dd,J=10.8,5.0 Hz,1H),2.76(d,J=16.4 Hz,1H),2.48(d,J=16.4 Hz,1H),2.03(s,1H),1.34(s,3H).13C NMR(100 MHz,CDCl3)δ170.93,136.68,128.14,128.02,125.68,123.67,115.96,68.66,40.17,39.48,22.57.HRMS(ESI)calcd for C11H14NO2[M+H]+192.101 9,found 192.101 4.
6-溴-4-(羟甲基)-4-甲基-3,4-二氢喹啉-2(1H)-酮(3f-3h):淡黄色固体,m.p.249~250 ℃;1H NMR(400 MHz,DMSO-d6)δ10.18(s,1H),7.39(S,1H),7.32(d,J=8.2 Hz,1H),6.80(d,J=8.3 Hz,1H),5.01(d,J=4.5 Hz,1H),3.44(dd,J=10.7,5.4 Hz,1H),3.29(dd,J=10.5,5.0 Hz,1H),2.50(d,J=15.9 Hz,1H),2.27(d,J=16.1 Hz,1H),1.15(s,3H).13C NMR(100 MHz,DMSO)δ169.56,137.67,131.94,130.40,128.92,117.65,114.26,67.34,40.04,39.60,22.84.HRMS(ESI)calcd for C11H13BrNO2[M+H]+270.012 4,found 270.012 3.
4-(羟甲基)-4-苯基-3,4-二氢喹啉-2(1H)-酮(3i-3j):淡黄色固体,m.p.204~205 ℃;1H NMR(400 MHz,DMSO-d6)δ10.01(s,1H),7.40(d,J=7.6 Hz,1H),7.21(m,6H),7.04(t,J=7.5 Hz,1H),6.89(d,J=7.8 Hz,1H),5.15(t,J=4.7 Hz,1H),4.01(dd,J=10.9,5.1 Hz,1H),3.74(dd,J=11.0,4.9 Hz,1H),2.93(s,2H).13C NMR(100 MHz,CDCl3)δ170.09,143.12,138.76,128.52,128.10,127.96,127.64,127.47,126.95,122.41,116.17,107.03,66.57,47.75.HRMS(ESI)calcd for C16H16NO2[M+H]+254.117 6,found 254.116 8.
2 结果与讨论
2.1 反应条件优化
选用化合物1a向目标化合物3a转化的反应作为优化反应条件的模板反应(图4(a)).实验表明,以1∶1的体积比例混合乙二醇(Glycol)和水作为反应溶剂(n1a∶nKOH=1∶2.1),回流反应6 h以后,产物3a的收率可以达到71%(表2,实验1).随着氢氧化钾用量的下降反应时间会显著延长,产物3a的产率也会明显地下降(表2,实验2和3).更重要的是,实验还表明乙二醇和水都是这一过程中必不可少的溶剂(表2,实验4和5).使用其他的碱,包括氢氧化钠(NaOH)、氢氧化钡(BaOH)和氢氧化锂(LiOH)等,均不能提高目标产物3a的产率(54%、58%和53%)(表2,实验6~8).另外,在80 ℃下实施的反应只能给出24%的3a(表2,实验9).其他醇类溶剂,包括甲醇(MeOH)、乙醇(EtOH)、正丁醇(nBuOH)和叔丁醇(tBuOH)在与水等比例混合后均不能提高反应效率,产物3a的收率都不超过46%(表2,实验10~13).筛选实验表明,最优的反应条件是使用氢氧化钾(n1a∶nKOH=1∶2.1),3 mL体积比为1∶1的乙二醇和水混合溶剂,加热回流(表2,实验1).
2.2 反应底物扩展
确定了最优的反应条件以后(表2,实验1),对该分子内重排扩环合成3,4-二氢喹啉-2(1H)-酮3反应的适用范围进行了探索,主要结果列于表3.实验表明,对于苯环上不带有任何取代基和C7位带有溴取代基的呋喃[3,2-b]吲哚类底物1(a-e)和1(f-h),反应对各种酰基保护基,包括:乙酰基、一氯乙酰基、三氯乙酰基、苯甲酰基和特戊酰基都表现了良好的兼容性.所有反应均在6 h内完成并以68%~75%的产率生成产物3(a-h).空间位阻没有影响反应的效率,因为8b位置带有苯基的底物1i和1j也分别以71%和69%收率生成了目标产物3i和3j.然而,对于苯环部分带有烷氧基的化合物1k和1l,反应生成了复杂的化合物.C8b位置没有甲基取代的底物1m表现出了类似的反应活性.值得一提的是,对于对甲苯磺酰基(Ts)取代的底物1n,该反应不能发生,90%的原料被回收.由于方法学上底物来源的限制,没有进一步扩展该反应底物的适用范围[36].
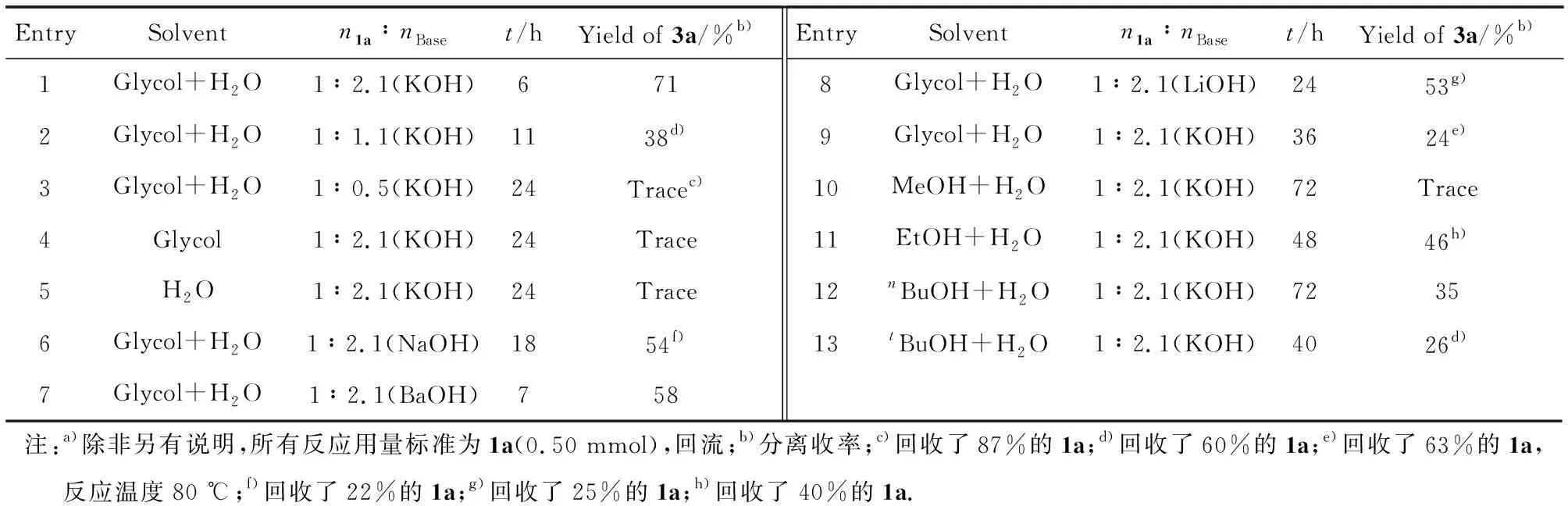
表2 反应条件筛选a)
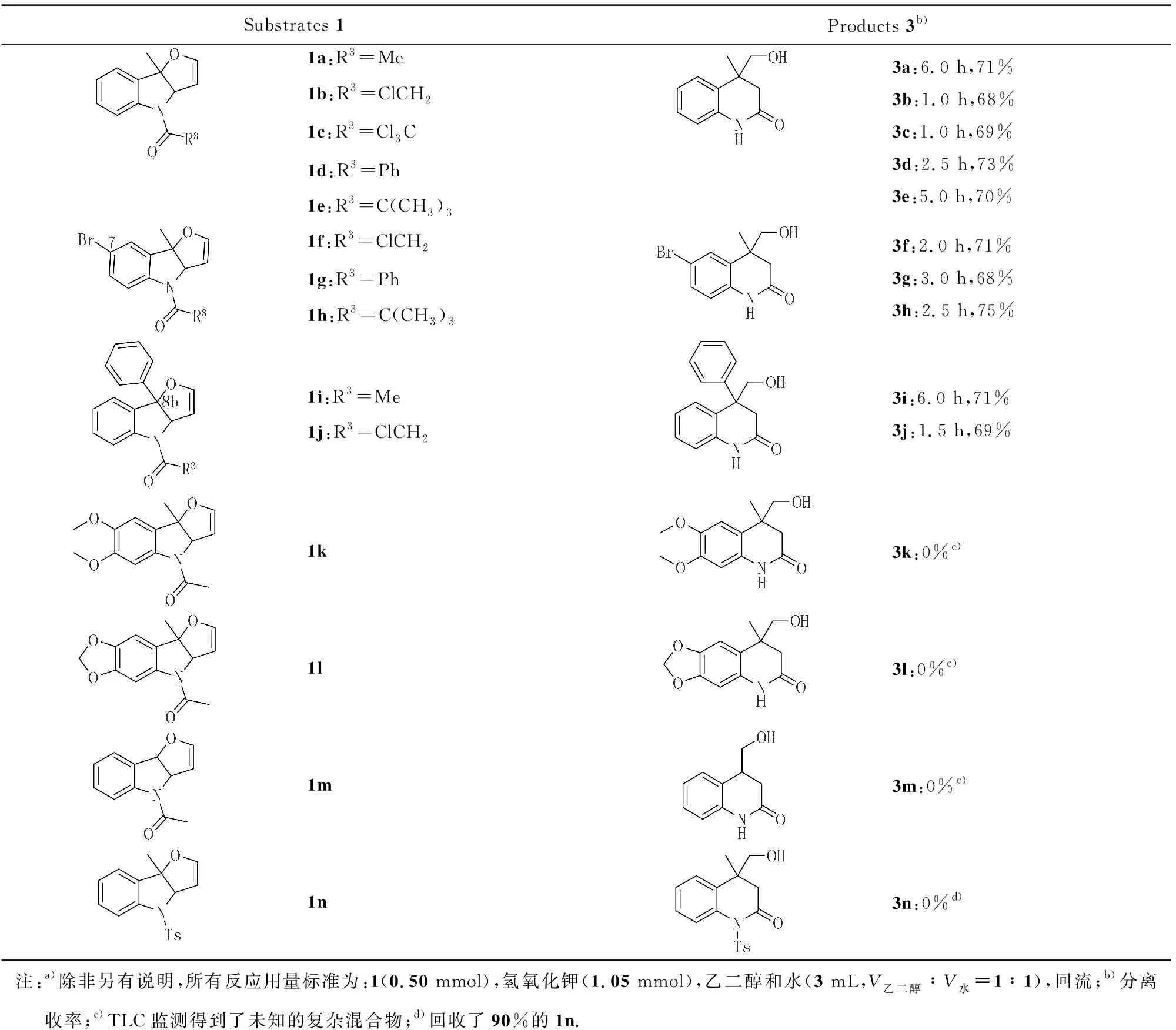
表3 底物扩展a)
3 结 论
本文发展了一种碱性条件下含有吲哚结构单元的多并杂环类化合物的重排反应,合成了一系列重要的亚甲羟基取代的喹啉酮类化合物.由于通过控制实验没有捕捉到反应的中间体,所以目前反应的具体机制尚不清楚.希望相关学者能够一起研究该反应的机制,这对于该碱性条件下吲哚类化合物的重排策略实现在有机合成化学中的应用甚至天然产物的人工合成方面非常重要.
