巴夫杜氏藻不同生长时期的转录组分析
2022-02-25宋韡汤丽群高帆
宋韡,汤丽群,高帆*
1(山西体育职业学院,山西 太原,030006)2(山西大学 生命科学学院,山西 太原,030006)
巴夫杜氏藻(Dunaliellaparva)属于绿藻门(Chlorophyta),绿藻纲(Chlorophyceae),团藻目(Volvocales),杜氏藻科(Dunaliellaceae),杜氏藻属(Dunaliella)[1],多分布于盐湖、海洋及湿地,为无细胞壁的单细胞真核生物,可进行无性或有性生殖。其含有丰富的生物活性物质和藻类色素,如多糖、甘油、β-胡萝卜素等,在保健品、食品加工、医疗和生物柴油等方面具有广阔的应用前景[2]。
目前,国内外对于巴夫杜氏藻的研究多集中于培养条件优化[3]、关键基因克隆分析[4]、活性物质提取等方面[5],对于其转录组学方面的研究很少。进入21世纪以来,随着国内外高通量测序技术快速发展,以Ion Torrent、Roche 454、Thermo Fisher、SOLiD、HiSeq为代表的测序平台[6]被广泛使用。该技术已成为在不同生长阶段、不同胁迫处理等条件下获取物种关键功能基因,预测代谢通路及表征生物学过程的重要手段之一[7]。YAO等[8]对Dunaliellatertiolecta进行转录组测序,获得10 185条与脂肪酸合成相关的基因。PUENTE-SNCHEZ等[9]对Dunaliellaacidophila进行测序,筛选出不同重金属胁迫下的关键基因。HE等[10]对D.salina在高盐度胁迫下的转录组数据进行分析,获取了耐盐关键基因及其代谢通路。上述转录组测序分析均基于杜氏藻属的其他种类,对D.parva的研究还仅限于2016年尚常花对限氮条件下巴夫杜氏藻的转录组和蛋白组测序分析[11]。该研究通过Illumina HiSeq 2000测序平台预测了D.parva油脂合成途径,鉴定了油脂合成关键基因wri1和Lip,但并未对其不同生长时期的转录水平及相关基因进行分析。BGISEQ-500是新一代高通量测序平台,利用该平台进行转录组测序研究已在多个物种中报道[12]。本研究利用该测序平台对不同生长时期巴夫杜氏藻进行转录组测序分析,获得大量差异表达基因,并预测其功能及参与的代谢通路。研究结果为该藻生长发育调控机制及相关代谢途径的研究奠定了基础,对该藻类资源改良与分子选育具有重要的指导意义。
1 材料与方法
1.1 藻种收集与培养
巴夫杜氏藻种(D.parva),英国CCAP藻种库,藻细胞活化后用优化杜氏盐藻培养基进行光照静态培养,培养基配方:FeC6H5O70.005 g,NaH2PO4·2H2O 0.015 g,CaCl2·2H2O 0.045 g,KCl 0.075 g,NaNO30.45 g,NaHCO30.85 g, MgSO4·7H2O 1.25 g,NaCl 130 g,Co(NO3)2·6H2O 5 mg,CuSO4·5H2O 8 mg,ZnSO4·7H2O 23 mg,维生素B10.5 mg,pH调至7.5,ddH2O定容至1 000 mL。光照强度20 000 lx,温度(25±5) ℃,光周期16 h∶8 h,培养室保持持续无菌通风。每隔7 d采样进行显微镜检,每隔21 d进行新鲜培养基补充,在培养液总体积不变的前提下确保培养过程无污染。
1.2 生长周期测定
参考王婷等[13]对杜氏藻生理指标的研究方法,利用紫外-可见光分光光度计测定685 nm处不同培养时间内的巴夫杜氏藻培养液吸光值。根据OD685数值大小,绘制巴夫杜氏藻生长周期曲线,测定其生长初期(包括停滞期和对数期)、中期(稳定期)和后期(衰亡期)的时间范围。
1.3 RNA提取,cDNA文库构建及转录组测序
分别取3份上述不同生长时期(OD685平均值所在时期)的巴夫杜氏藻液各300 mL,4 ℃低温离心(1 500×g)后用液氮速冻并快速研磨,用TRIzol试剂盒分别提取藻株总RNA,Agilent 2100 Bioanalyzer对其质量和浓度进行评估,利用反转录试剂盒反转录为cDNA,SMART cDNA文库构建试剂盒对合格样本进行文库构建。构建好的文库在BGISEQ-500测序平台进行转录组测序,上机测序操作委托深圳华大基因完成。
1.4 测序数据分析及de novo组装
利用SOAPnuke[14]对测序数据进行过滤与筛选,具体流程是:删除5′和3′端含接头及poly-N(N>5%)的低质量reads,筛选获得的Clean reads(Q20值> 95%,Q30值> 85%),利用Trinity软件对Clean reads进行从头组装。利用Bowtie2软件[15]预测Clean reads中的UniGenes,利用Tgicl软件[16]分析UniGenes数量和长度,利用BUSCO软件[17]评估组装质量。
1.5 转录组数据注释
基于转录组UniGenes与Gene Ontology(GO)、RefSeq Nonredundant Protein(NR)、Nucleotide Sequence(NT)、SwissPort、Pfam、Kyoto Encyclopedia of Genes and Genomes(KEGG)及Eukaryotic Orthologous Group(KOG)数据库数据的比对相似度(参数设置:E值≤1.00E-10,查询序列长度百分比≥85%, 无冗余Contigs)。基于Pfam和KOG数据库,利用HMMER程序[18]预测UniGene潜在功能及抗病毒结构域。
1.6 基因表达分析
利用Fragments per Kilobase of Transcript per Million Fragments(FPKM)[19]测算UniGene表达量,利用RSEM软件[20]测算差异表达基因(differential expression gene,DEG),利用DEseq2软件构建DEGs分布火山图(log2fold change≥|2.00|,Q值≤ 0.05),利用R语言构建不同样本的DEGs聚类热图。
1.7 GO和KEGG通路富集
利用Blast2GO[21]对UniGene进行GO条目分配和Ontology分类,基于R语言的Phyper功能对GO条目进行富集[错误发现率(false discovery rate,FDR)≤0.01]。利用KEGG通路的KAAS程序对UniGene进行KO条目分配和通路分类,基于R语言的Phyper功能对KEGG通路进行富集(FDR≤0.01)。
2 结果与分析
2.1 巴夫杜氏藻生长周期鉴定
基于巴夫杜氏藻的3个生物学重复生长曲线拟合,研究发现其生长周期大体可分为3个明显不同的阶段,即生长初期(含延滞期和对数期)、生长中期(成熟期或稳定期)和生长末期(衰亡期或凋亡期)。由图1-a可知,生长初期的平均OD685值为0.49±0.03,周期为0~45 d;生长中期的平均OD685值约为1.63±0.02,周期约为45~55 d;生长末期的平均OD685值为1.59±0.02,周期约为55~65 d。由图1-b可知,细胞色素在生长初期积累较少,整体呈浅绿色,藻细胞生长速度较快,细胞呈小球状;细胞色素在生长中期积累较多,整体呈深绿色,藻细胞呈椭球状,生长速度趋于稳定;细胞色素在生长末期的积累更深,整体呈墨绿色,藻细胞呈扁球状,生长速度趋于延滞。
2.2 RNA测序与de novo组装
巴夫杜氏藻RNA平均质量浓度为56 ng/μL,RNA完整值(RNA integrity number,RIN)平均值为7.1,符合RNA测序标准(RNA质量浓度≥ 30 ng/μL,RIN ≥ 6.0)。3个生长时期(30、50、60 d)共9个生物样本的原始测序数据共18.89 Gb,其中原始Reads约665.7 Mb,平均每个样本约71.76 Mb的Clean reads(总占比约72.23%)(表1)。共鉴定90 153条UniGene,检测到272条完整的Contig片段(完整度占比约90.23%,总长约7.65×107bp,平均长度约812 bp)(图2-a,表1)。N50长1 305 bp(N70长783 bp,N90长345 bp),GC占比约52.32%。UniGene中共预测到49 643条CDS(总长为3.90×107bp,N50为982 bp,GC占比53.65%),23 528条SSR(总长为20 412 bp)。12个测序样本的Q20和Q30百分比平均值分别为98.56%和89.43%。denovo组装的BUSCO质量评估结果显示,组装完整的测序片段、单拷贝及双拷贝片段总占比>90%(图2-b),组装质量较高。
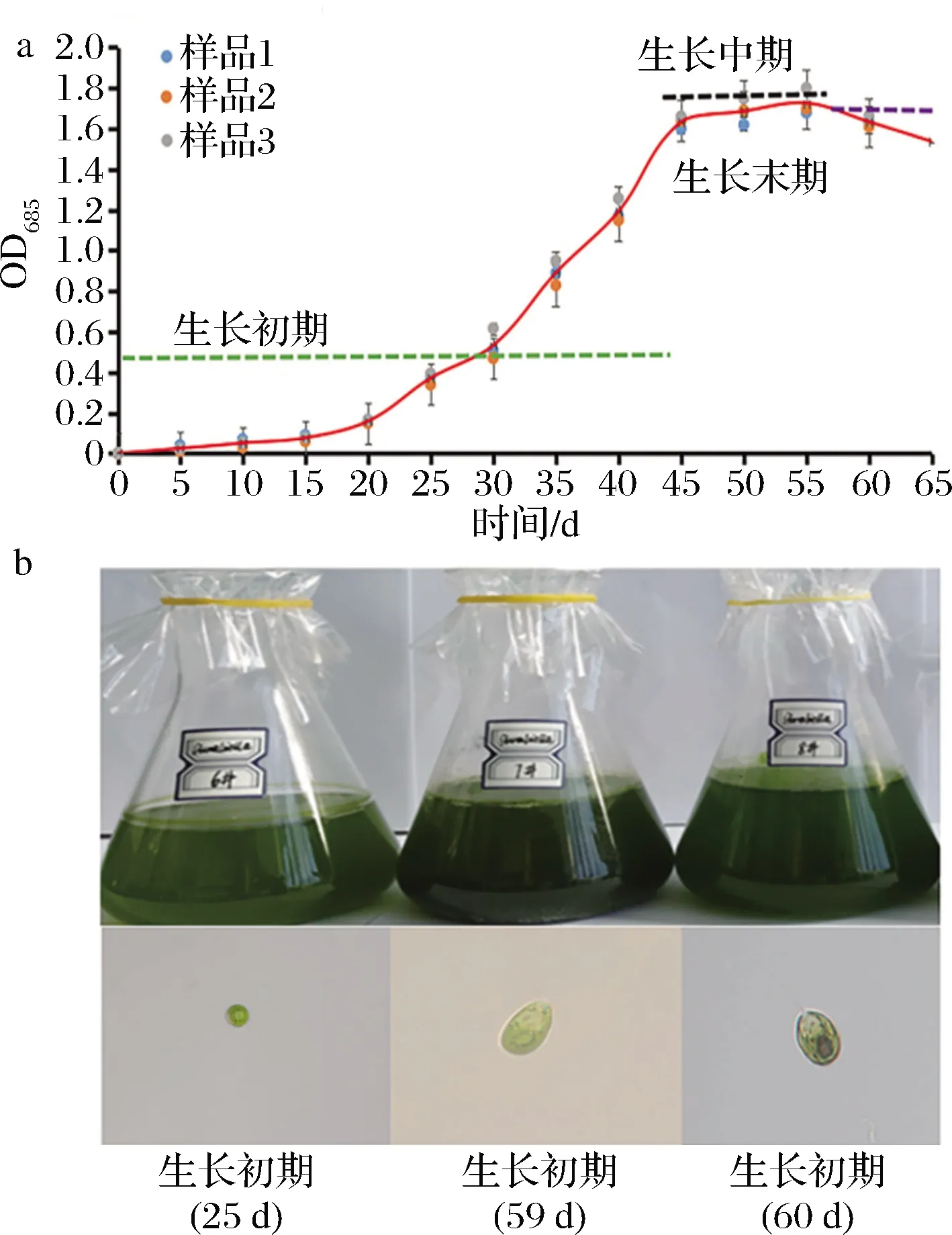
a-巴夫杜氏藻生长周期;b-巴夫杜氏藻形态学观察(标尺20 μm)图1 巴夫杜氏藻生长周期及其细胞形态Fig.1 Growth cycle and cell morphology of D. parva
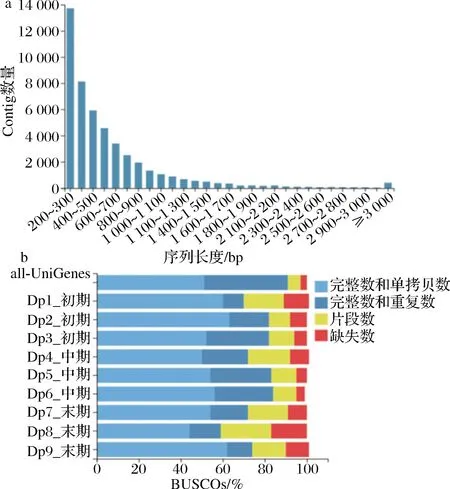
a-Contig长度分布;b-基于BUSCOs的de novo组装质量评估图2 转录组测序Contig长度及de novo组装质量分析Fig.2 Analysis of transcriptomic Contig length and de novo assembly quality
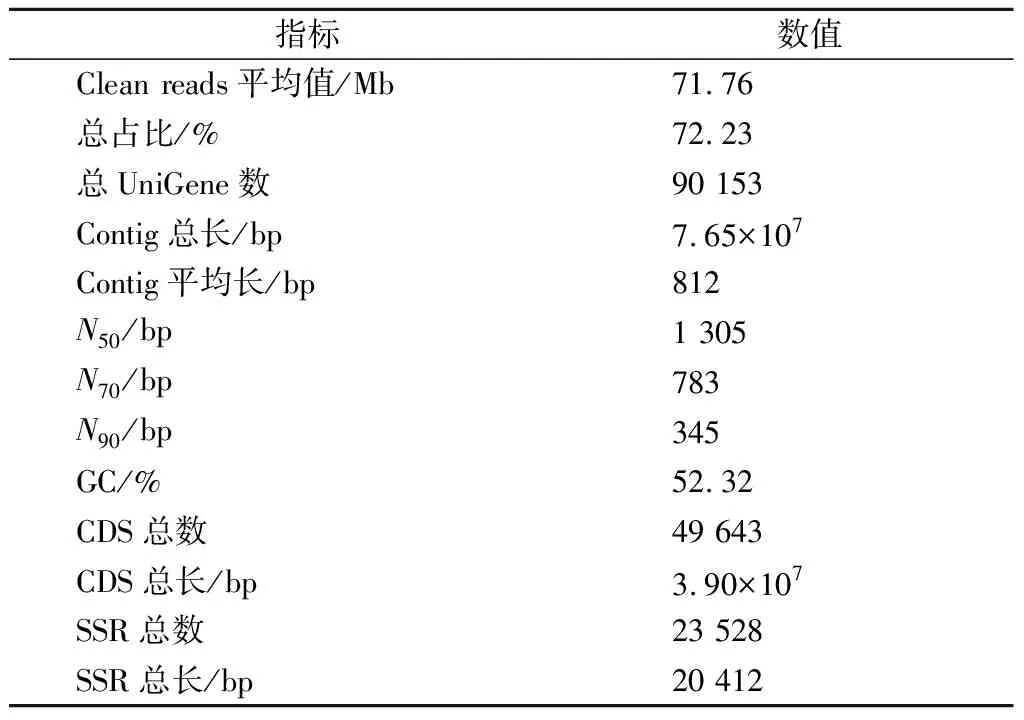
表1 巴夫杜氏藻转录组测序de novo组装基本信息Table 1 D.parva transcriptome de novo assembly information
2.3 基因注释与功能预测
90 153条UniGenes中平均约66.36%的基因可在七大生物信息数据库注释到(表2),其中约5.10%的基因在数据库中均有注释信息。每四大数据库的UniGene功能注释数详见图3。基于NR的UniGne注释物种分类结果显示,约23.43%的UniGene可在藻类(Chlamydomonaseustigma,Goniumpectorale和Volvoxcarter)和陆生植物(Dorcocerashygrometricum和Ricinuscommunis)中注释到,剩余76.57%的数据未在已报道物种基因信息库中检测到注释信息(图4-a)。注释信息最多的812条UniGenes中可预测到可变的N端病毒结构域,其次为510条UniGenes中预测到的受体样蛋白(receptor-like protein,RLP)结构域,仅仅3条UniGnes功能被预测与mol-like结构域形成相关(图4-b)。最多的4 650条UniGenes在KOG数据库中的功能注释为一般性调控功能,3 300条UniGenes的功能与后转录调控与蛋白质周转有关,最少的UniGne功能被预测与细胞运动与硫的形成相关(图4-c)。
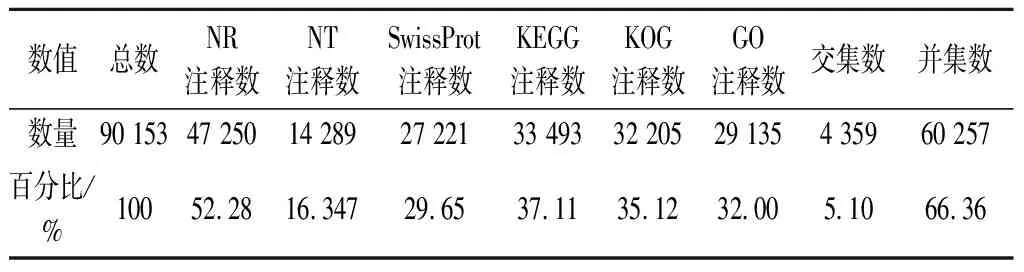
表2 巴夫杜氏藻的UniGene注释信息Table 2 UniGene annotation of D.parva
2.4 基因表达与聚类分析
由图5-a可知,基于UniGene表达FPKM值,巴夫杜氏藻生长中期和末期的基因整体可聚为一类,具有相似的表达趋势,生长初期的基因单独聚为一类,所有UniGene大致可划分为四大簇(Cluster Ⅰ、Ⅱ、Ⅲ和Ⅳ)。以巴夫杜氏藻生长初期UniGene为对照,共检测到86 387条差异表达基因(图5-b和图5-c)。相对于生长初期,生长中期共检测到上调基因14 556条,下调基因22 589条;生长末期共检测到上调基因18 940条,下调基因30 302条。两对比较组间共有差异表达基因有20 579条(图5-d)。
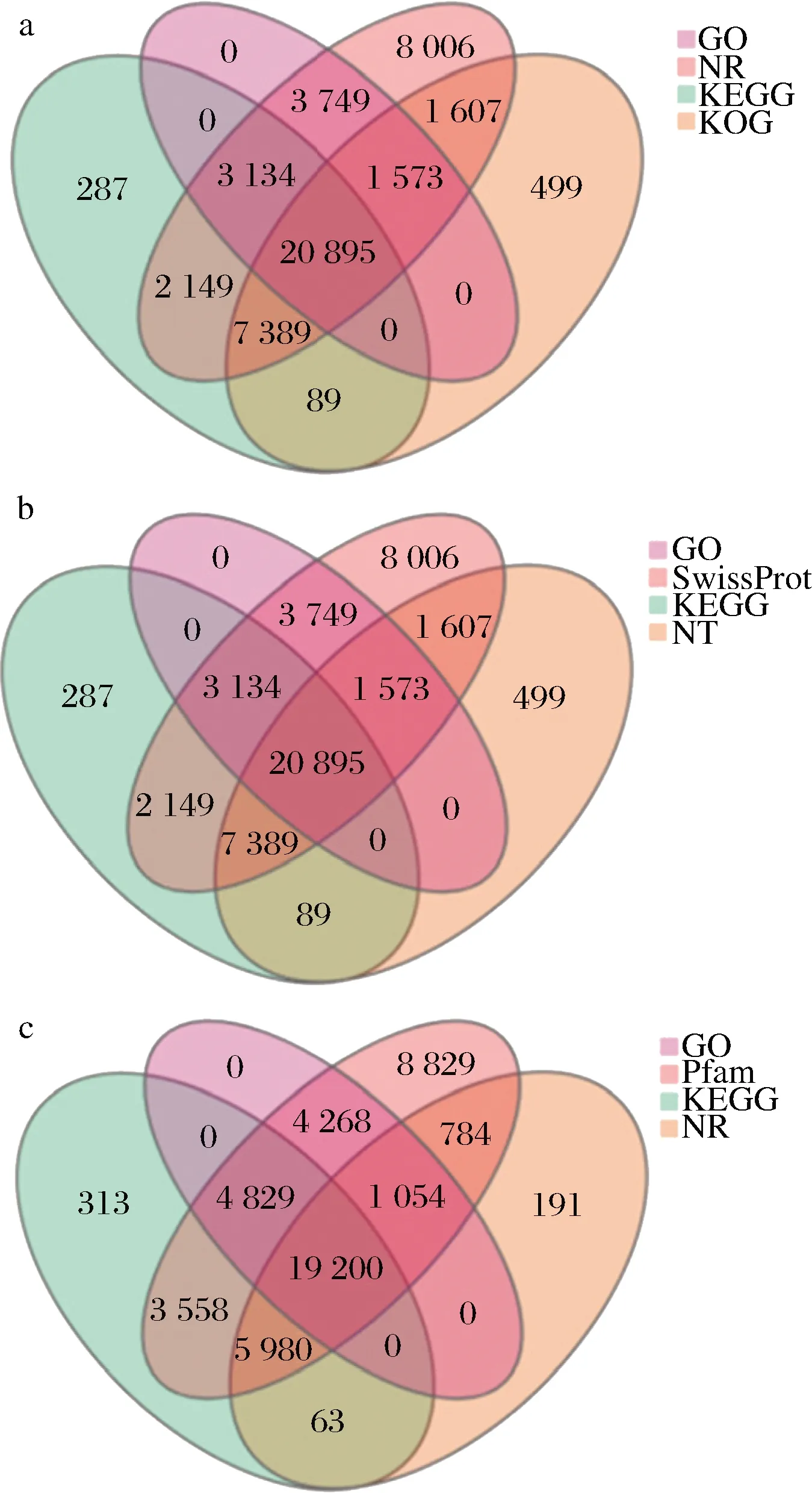
a-GO-NR-KEGG-KOG注释维恩图;b-GO-SwissProt-KEGG-NT 注释维恩图;c-GO-Pfam-KEGG-NR注释维恩图图3 巴夫杜氏藻unigene注释维恩图Fig.3 Venn diagram of D.parva unigenes
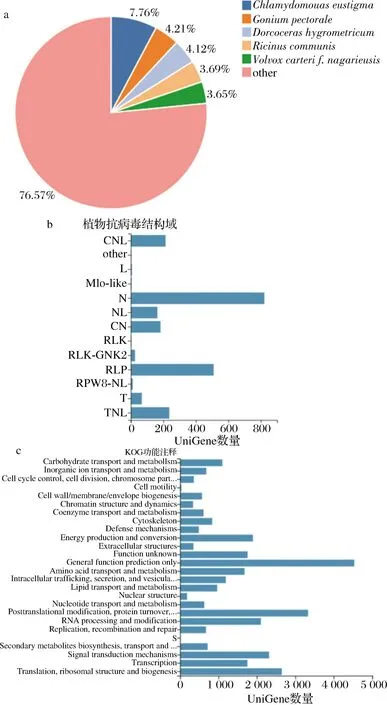
a-巴夫杜氏藻UniGene物种分类注释;b-巴夫杜氏藻UniGene 抗病毒基因结构域注释;c-巴夫杜氏藻UniGene KOG功能注释图4 巴夫杜氏藻UniGene注释Fig.4 UniGene annotation of D.parva
2.5 GO分类与富集
由图6-a和图6-b可知,在生物学过程类别中,GO条目最多的是细胞过程(生长中期vs.生长初期的基因数为2 250,生长末期vs.生长初期的基因数为6 300);在细胞组分类别中,GO条目最多的是细胞(生长中期vs.生长初期的基因数为1 968,生长末期vs.生长初期的基因数为4 987);在分子功能类别中,GO条目最多的是催化活性(生长中期vs.生长初期间的基因数为2 419,生长末期vs.生长初期的基因数为7 962)。由两对巴夫杜氏藻比较组的差异基因GO富集top 20结果可知(图6-c和图6-d),富集程度最高的GO条目均是膜的整体成分,生长中期vs.生长初期的基因数最高为1 140,富集比值最高为0.42;生长末期vs.生长初期的基因数最高为3 469,富集比值最高为0.41。
2.6 KEGG通路分类与富集
由图7-a和图7-b可知,2对比较组间所有的通路均可划分为细胞过程、环境信息处理、遗传信息处理、新陈代谢和有机体系统。其中,通路基因条目最多的是整体代谢通路(生长中期vs.生长初期的基因数为1 200,生长末期vs.生长初期的基因数为4 300)。由两对巴夫杜氏藻比较组的差异基因KEGG通路富集top 20结果可知(图7-c和图7-d),生长中期vs.生长初期富集程度最高的通路是剪接体(基因数为546,富集比值为0.16);生长末期vs.生长初期富集程度最高的通路是RNA转运(基因数最高为1 521,富集比值最高为0.15)。
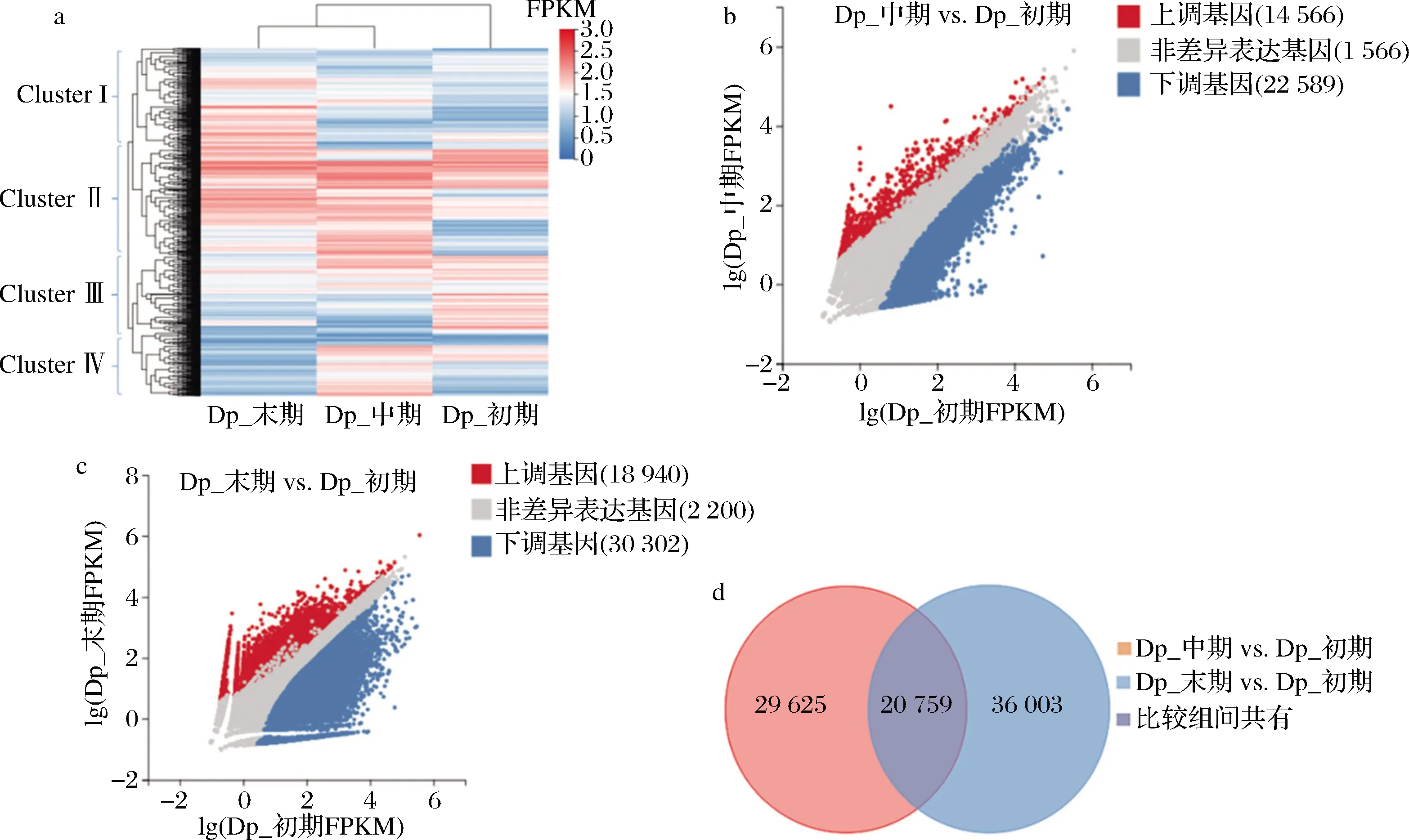
a-不同生长时期的基因表达聚类热图;b-生长中期与生长初期间差异基因表达散点图;c-生长末期与生长初期间差异基因 表达散点图;d-两对比较组间的共有差异基因维恩图图5 巴夫杜氏藻不同比较组间的差异表达基因分析Fig.5 Analysis of differentially expressed genes among different D.parva comparison groups
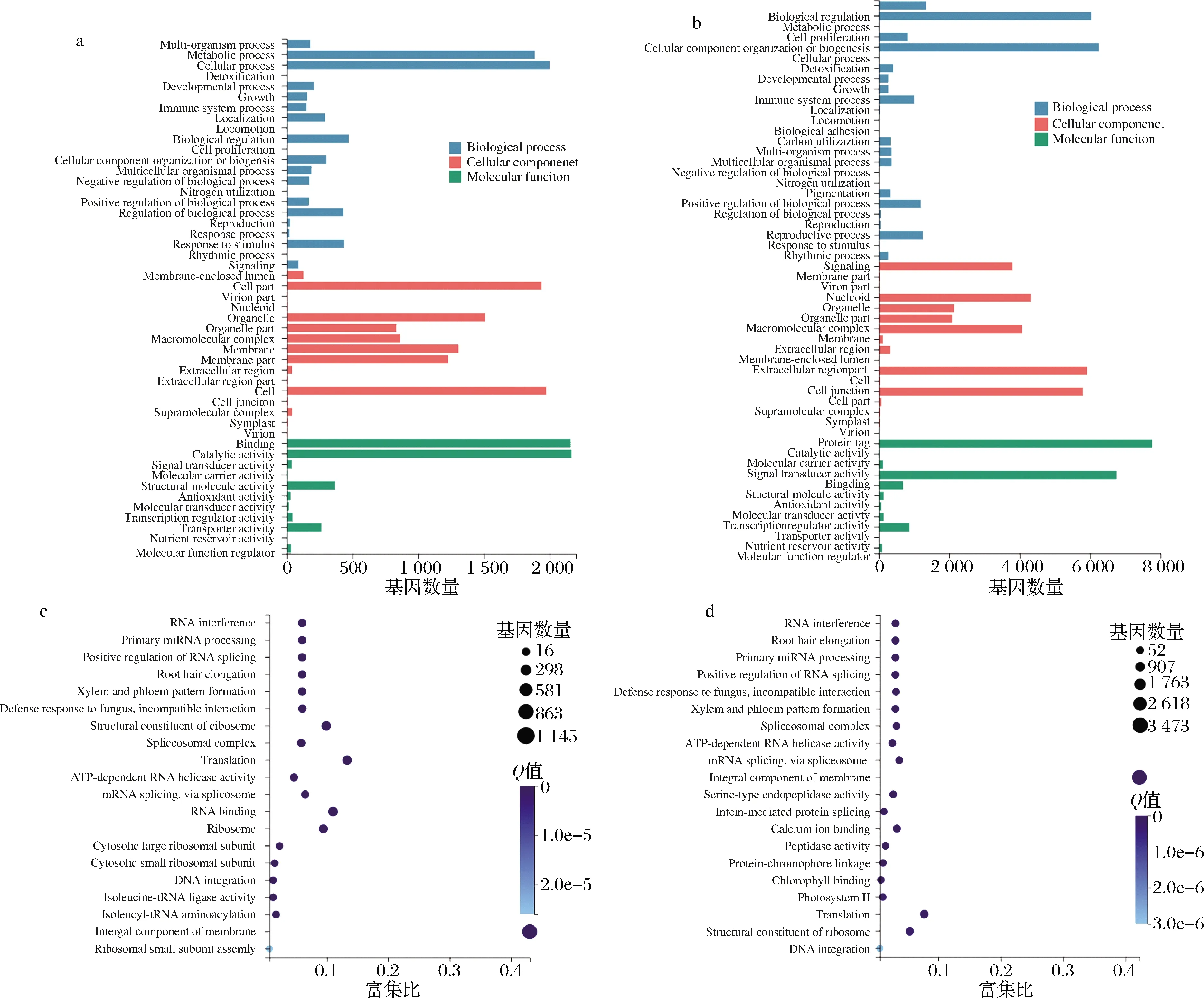
a-生长中期与生长初期间差异基因GO分类;b-生长末期与生长初期间差异基因GO分类;c-生长中期与生长初期间差异基因 GO富集;d-生长末期与生长初期间差异基因GO富集图6 巴夫杜氏藻不同比较组间的差异基因GO分类与富集Fig.6 GO classification and enrichment of DEGs among different D.parva comparison groups
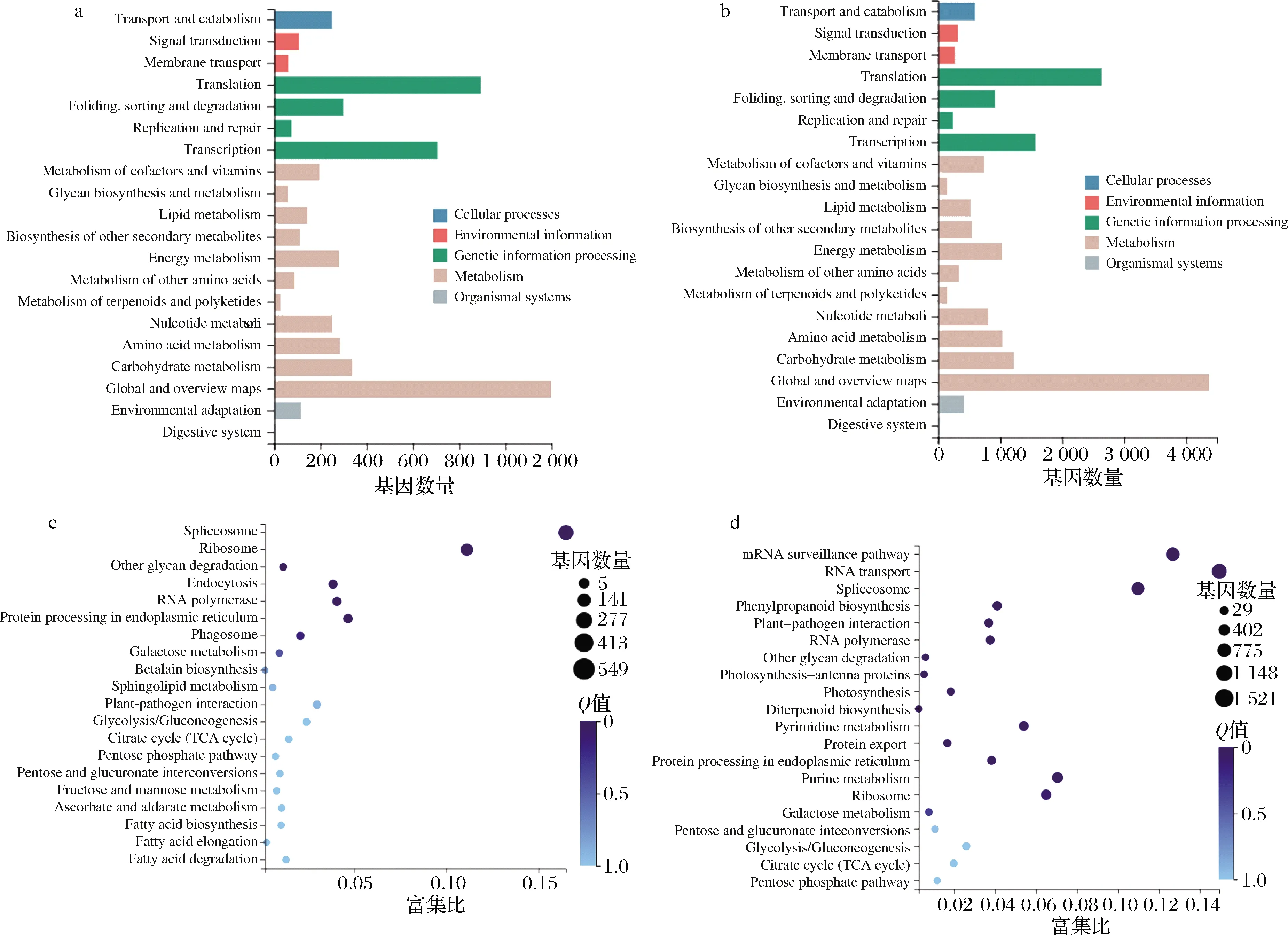
a-生长中期与生长初期间差异基因KEGG通路分类;b-生长末期与生长初期间差异基因KEGG通路分类;c-生长中期与生长初期间 差异基因KEGG通路富集;d-生长末期与生长初期间差异基因KEGG通路富集图7 巴夫杜氏藻不同比较组间的差异基因KEGG通路分类与富集Fig.7 KEGG pathway classification and enrichment of DEGs among different D.parva comparison groups
3 讨论
目前,转录组测序技术已成为阐明藻类功能基因的重要手段之一。本研究通过第二代高通量测序平台BGISEQ-500对不同生长时期的巴夫杜氏藻进行转录组测序,获得的单样本Clean reads(71.76 Mb)及GC百分比(52.32%)均高于PANAHI等[22]利用Illumina GAIIx测序平台获得的盐生杜氏藻(D.salina)数据(Clean reads 31.2 Mb,GC百分比50%)。巴夫杜氏藻cDNA测序文库的Q20和Q30平均值高于POLLE等[23]报道的D.salinaCCAP 19/18转录组测序的Q20和Q30值。但本研究预测的CDS数(49 643条)却低于HE等[10]在Illumina HiSeq 2 000测序平台获得的D.salinaCCAP 19/30CDS数(49 751条)。巴夫杜氏藻转录组测序数据denovo组装后的N50长度(1 305 bp)小于HONG等[7]从D.salina435转录组测序获得的N50长度(1 727 bp)。由此可知,基于BGISEQ-500测序平台获取的巴夫杜氏藻转录组测序数据整体是可靠的。
仅23.43%的巴夫杜氏藻UniGene可在植物物种中注释到,相较于其他藻类物种注释信息,比例较低(一般>40%)[24]。笔者推测其原因可能是:(1)该情况可能与巴夫杜氏藻原始系统进化地位相关。巴夫杜氏藻是绿藻的一种,研究显示绿藻起源较早,相比高等陆生植物而言,其与蓝细菌甚至与某些原生动物系统进化关系较近[25],而本研究未将UniGene的物种注释范围扩大到细菌及原生动物。(2)本研究测序获得UniGene数据未经开放型阅读框(open reading box,ORF)预测处理,由此冗余数据及非功能基因的片段对物种注释信息造成了干扰。因此,在原UniGene数据基础上,扩大其物种基因注释的数据库范围,并进一步处理搜集含ORF框的功能基因数据,将可能提高其基因的物种注释比例。
以生长初期为对照,巴夫杜氏藻生长中期差异表达基因数低于生长末期,这表明到了藻类的生长末期,随着细胞的凋亡、代谢废物及有毒物质的积累,用于调控生长危机的基因数明显增加,以此应答不利于藻类营养物质积累及细胞增殖的细胞内环境[26]。此外,相对于生长初期,巴夫杜氏藻生长中期和生长末期的下调基因数均高于上调基因数,这暗示了随着生长周期延长,对藻株生长发育和新陈代谢等生物学过程起重要作用的基因来说,其总体调控方式是负向的,这与杜氏藻在不同非生物胁迫下的差异基因统计结果有所差异(一般上调基因数高于下调基因)[8-10]。
不同比较组间基因GO条目与功能富集多数与细胞的完整性及其功能组分相关,这表明随着生长周期延长,巴夫杜氏藻胞内的甘油、多糖、β-胡萝卜素等生物活性内含物逐渐积累,细胞体积不断增大[27],相关基因的功能多与细胞周期调控、细胞组分合成及催化活性紧密相关。不同比较组间基因参与的KEGG通路与多种代谢过程相关,但其详细的代谢途径仍不明确。究其原因,可能与已报道藻类代谢通路信息较少有关。
4 结论
不同生长时期巴夫杜氏藻转录组测序结果及组装质量较高,上调表达基因数高于下调表达基因,GO富集条目最多的是细胞过程、细胞整体成分及催化活性,KEGG通路富集程度最高的是剪接体及RNA转运代谢通路。下一步,对该藻株生长发育关键基因的挖掘及其表达验证,蛋白组与代谢组学的联合分析其参与的关键代谢通路很有必要,这对于深入揭示巴夫杜氏藻的生长发育分子机制,分子水平调控其活性物质含量,遗传水平改良其资源品质具有重要的意义。
