反义寡核苷酸技术在遗传性视网膜变性治疗中的应用
2022-02-23李五一综述睢瑞芳审校
李五一 综述 睢瑞芳 审校
中国医学科学院 北京协和医学院 北京协和医院眼科 100730
长期以来,遗传性视网膜变性缺乏有效的临床治疗手段。近20年中,以腺相关病毒(adeno-associated virus,AAV)和慢病毒为载体的基因增补策略在遗传性视网膜变性治疗研究中已取得重大进展,为遗传性视网膜变性治疗带来了希望,但由于载体的负载基因长度存在局限性,使其难以广泛应用。反义寡核苷酸(antisense oligonucleotide,AON)是一种通过碱基互补配对原则结合并降解靶信使RNA(messenger RNA,mRNA)以纠正或抑制其转录与翻译的小分子化合物。AON具有特异性高、微量高效、免疫原性低、毒性及不良反应小、应用范围广等优点,可以有效地填补遗传性视网膜变性基因治疗的空白。AON已在遗传性视网膜变性和视神经萎缩等眼部疾病治疗研究中取得了一定的进展,本文就近年来AON在其化学结构修饰、特性和作用机制等方面的进展及其在不同遗传性视网膜变性疾病中应用的治疗策略进行综述。
1 AON作用机制和结构修饰
1.1 AON作用机制
AON通过碱基配对原理与靶mRNA上特定序列结合,活化核糖核酸酶H(ribonuclease H,RNase H),使靶mRNA降解以抑制或调控其转录、剪切、翻译,从而调节其编码蛋白的表达[1]。人体细胞内包含有2种RNase H酶,即RNase H1和RNase H2,其中RNase H2表达更为丰富,但仅RNase H1参与AON的作用机制[2-3]。当DNA与RNA形成异源双链体时,RNase H酶被激活并诱导靶mRNA降解,而AON在诱导双链体中mRNA分解后,可以继续特异性结合其他靶mRNA以持续发挥作用[4]。AON可以通过多种不同的机制作用于靶mRNA翻译为蛋白质的一系列过程,进而发展出了多种AON应用方式[5-6](图1),为其在遗传性疾病临床治疗中的应用奠定了基础。

图1 AON的多种作用机制图 绿色双链代表DNA;蓝绿色单链代表由DNA转录的RNA;红色小片段代表AON;AON:反义寡核苷酸;mRNA:信使RNA
AON可以与前体mRNA 5’-端非翻译区(5’-untranslated region,5’-UTR)结合抑制其甲基化,或者结合3’-端抑制其多聚腺苷化,从而导致mRNA不稳定而分解,达到等位基因特异性抑制的作用[2,7]。对于成熟的mRNA,AON可以通过结合起始密码子的方式形成mRNA的翻译阻滞,从而起到抑制蛋白表达的作用[7]。在前体mRNA内含子去除及外显子连接的过程中,AON可以通过靶向内含子和外显子之间的剪接位点或其周围的增强子及沉默子位点,纠正前体mRNA的异常剪切,达到等位基因特异性修复的目的[4,8]。同时,AON干预剪切的能力也可用于调控前体mRNA的治疗策略,这种方式被称为外显子跳跃,指当突变导致阅读框改变、异常剪切产生假外显子或所翻译的蛋白质功能受损时,通过AON靶向剪接位点以外显子跳跃的方式恢复全部或大部分蛋白功能[9],这是其靶向治疗遗传性疾病中的常用治疗策略。Echevarría等[10]在对欧美地区杜氏肌肉萎缩症(Duchenne muscular dystrophy,DMD)患者的基因治疗研究中发现,突变导致的外显子50的跳跃造成阅读框提前结束并产生了截短的非功能性蛋白;通过设计AON结合外显子51的剪切元件并跳过外显子51的方式修复了mRNA的阅读框,并产生了较短的功能性蛋白,用于治疗突变导致外显子50跳跃的药物Exondys51(eteplirsen,依特立生)可惠及美国13%的DMD患者。
1.2 AON的结构修饰
AON是由磷酸二酯键连接而成的线性核苷酸片段,长度为13~25 bp。AON在细胞内会因被核酸外切酶和内切酶迅速降解而无法发挥其药物作用,限制了其在遗传性疾病临床中的应用,因此需要对其结构进行化学修饰以改变其特性[11]。根据AON的分子结构,可以通过3个方面对其进行修饰,即碱基、核糖和磷酸二酯键(图2,表1)。

图2 AON的化学结构修饰示意图 核糖2’位置常用的第1、2、3代化学修饰 PS:硫代磷酸酯;2’-OMe:2’-氧-甲氧基;2’-MOE:2’-氧-甲氧乙基;LNA:锁核酸;PMO:磷酰二胺吗啉代低聚物;PNA:肽核酸
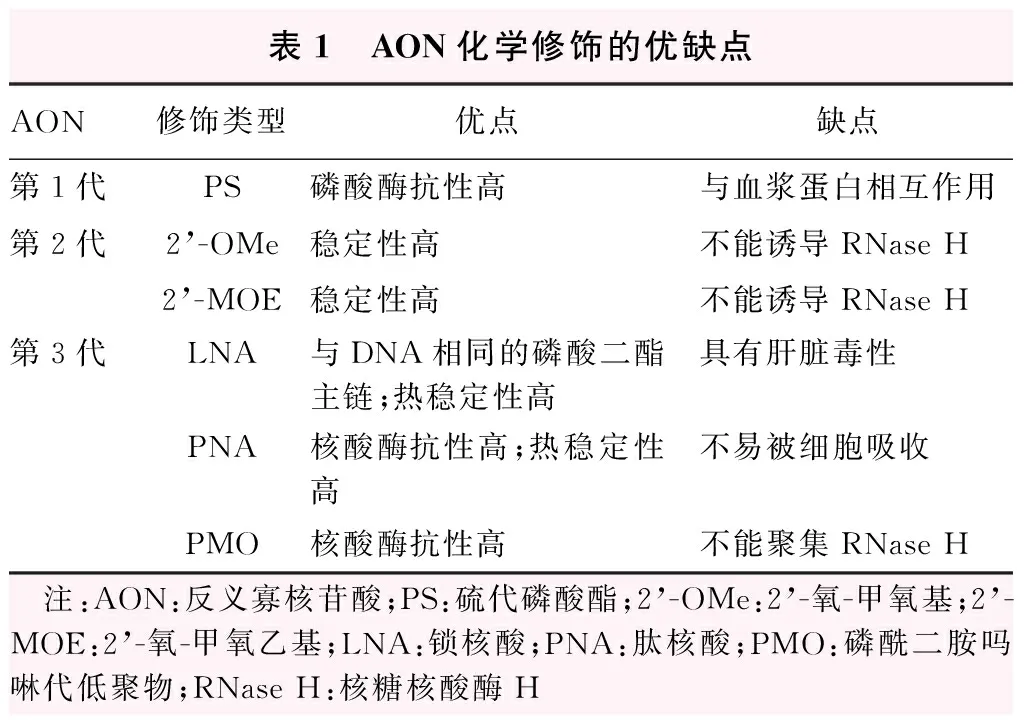
表1 AON化学修饰的优缺点AON修饰类型优点缺点第1代PS磷酸酶抗性高与血浆蛋白相互作用第2代2’-OMe稳定性高不能诱导RNase H2’-MOE稳定性高不能诱导RNase H第3代LNA与DNA相同的磷酸二酯主链;热稳定性高具有肝脏毒性PNA核酸酶抗性高;热稳定性高不易被细胞吸收PMO核酸酶抗性高不能聚集RNase H 注:AON:反义寡核苷酸;PS:硫代磷酸酯;2’-OMe:2’-氧-甲氧基;2’-MOE:2’-氧-甲氧乙基;LNA:锁核酸;PNA:肽核酸;PMO:磷酰二胺吗啉代低聚物;RNase H:核糖核酸酶H
第1代AON的修饰主要针对寡聚脱氧核苷酸磷酸二酯键的分子骨架,利用硫原子取代磷酸骨架上的非成键氧原子,形成硫代磷酸酯AON(phosphorathioate antisense oligonucleotides,PS-AONs),以增强其对核酸酶降解作用的抵抗能力[12],提高RNase H对靶mRNA降解的活性[11],并且增强了AON的结构稳定性。但是PS-AONs的键序和电荷定位显示硫代磷酸根中硫原子带有负电荷,会导致PS-AONs及其核酸降解物与蛋白受体非特异性结合[13]。Watanabe等[14]研究结果表明,PS-AONs表现出与血浆蛋白结合的特性,并提出了AON的毒性问题。
第2代AON在PS-AONs的基础上对核糖结构进行修饰。通过将其核糖2’位置修饰为2’-氧-甲氧基(2’-O-methyluridine,2’-OMe)和2’-氧-甲氧乙基(2’-O-methoxyethyl,2’-MOE)的方式,解决了AON与蛋白非特异性结合的问题,减轻其毒性;同时进一步增强其核酸酶抗性,提高结构稳定性,并增加其与靶序列的亲和力[4,15]。RNase H的激活需要AON中含有5~7个连续的脱氧核苷酸片段,然而AON核糖2’位置的结构改变影响了RNase H的降解作用,进而降低了AON的有效性[5]。Gapmer是一种AON的嵌合形态,其中央序列纳入至少5个未修饰的脱氧核苷酸用于激活RNase H,进而克服了核糖结构修饰后AON有效性降低的缺陷[16]。由于核糖环修饰部分的长度与RNase H有效激活相关[17],最佳中央序列的长度取决于基序,但脱氧核苷酸序列过长会影响其与mRNA的结合力,因此建立gapmer结构时需要在序列长度和结合力之间保持平衡[16]。2’-MOE-PS gapmer AON和2’-OMe-PS gapmer AON是第2代AON中常用的修饰组合[4],也是目前遗传性疾病临床应用中常见的化学修饰类型。
第3代AON结合了磷酸酯骨架和核糖环的修饰,在提高核酸酶抗性、降低毒性及增加靶向结合力和有效性之间进行平衡和优化,增加了更多的结构变化。锁核酸(locked nucleic acid,LNA)、肽核酸(peptide nucleic acid,PNA)和磷酰二胺吗啉代低聚物(phosphorodiamidate morpholino oligomers,PMO)为3种具有代表性的特殊修饰结构。这些特殊修饰同样会影响AON对RNase H的激活,其中LNA可以通过gapmer形式形成嵌合AON,由中央序列保留的脱氧核苷酸序列激活RNase H,在提高核酸酶抗性的同时,有效提高RNase H对靶向序列的降解能力[5]。但LNA修饰的gapmer AON在动物实验中显示出明显的肝脏毒性,限制了其临床应用的开发[18]。这类新型修饰可不依赖RNase H的机制,通过靶向mRNA的5’非翻译区,阻止核糖体的组装,形成翻译阻滞机制[5,19]。LNA、PNA和PMO这3种AON修饰都具有很好的结构稳定性和靶向结合力,且均为不带电的分子,从而降低了其与蛋白非特异性反应的可能性,但呈电中性的骨架也降低了其溶解度和生物膜渗透性,影响了其递送和效能[4],导致其在临床治疗中的应用并不广泛。
2 AON在遗传性视网膜变性治疗中的应用
AON在遗传性眼病的治疗中有许多优势。由于眼球是一个相对独立的器官并拥有血-眼屏障,体积相对较小,AON的给药剂量较低,故可以在一定程度上减轻系统免疫反应。同时眼内注射给药安全性高、难度小,使AON在遗传性眼病的应用进展迅速。目前,已有7种针对遗传性眼病致病基因的AON药物,其中3种AON已进入临床试验阶段。迄今为止,AON治疗遗传性眼病的研究范围十分广泛,已应用3种不同化学修饰的AON对5种疾病的致病变异进行治疗,其中4种为遗传性视网膜变性疾病。根据不同的疾病类型和突变位点,可以应用AON的多种作用机制,如常染色体显性遗传的疾病可以通过等位基因特异性抑制的策略来减少其突变蛋白的表达,或者通过等位基因特异性修复的方法恢复其野生型蛋白的表达;常染色体隐性遗传的疾病则常通过外显子跳跃的方式跳过突变的位点以形成截短但具有功能性的蛋白,或者通过造成假外显子跳跃的策略恢复其正常蛋白的表达(表2)。
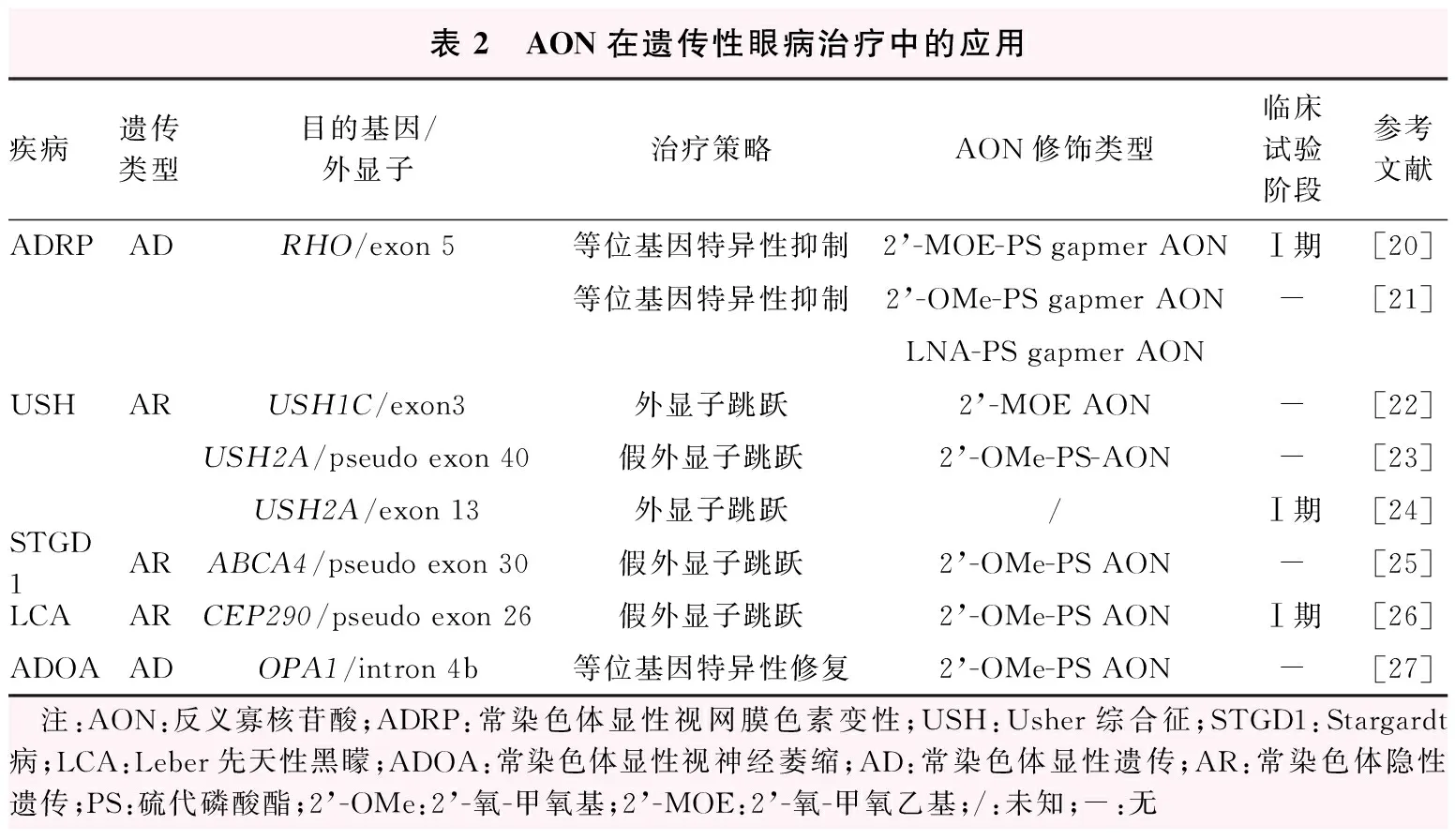
表2 AON在遗传性眼病治疗中的应用疾病遗传类型目的基因/外显子治疗策略AON修饰类型临床试验阶段参考文献ADRPADRHO/exon 5等位基因特异性抑制2’-MOE-PS gapmer AONⅠ期[20]等位基因特异性抑制2’-OMe-PS gapmer AON-[21]LNA-PS gapmer AONUSHARUSH1C/exon3外显子跳跃2’-MOE AON-[22]USH2A/pseudo exon 40假外显子跳跃2’-OMe-PS-AON-[23]USH2A/exon 13外显子跳跃/Ⅰ期[24]STGD1ARABCA4/pseudo exon 30假外显子跳跃2’-OMe-PS AON-[25]LCAARCEP290/pseudo exon 26假外显子跳跃2’-OMe-PS AONⅠ期[26]ADOAADOPA1/intron 4b等位基因特异性修复2’-OMe-PS AON-[27] 注:AON:反义寡核苷酸;ADRP:常染色体显性视网膜色素变性;USH:Usher综合征;STGD1:Stargardt病;LCA:Leber先天性黑矇;ADOA:常染色体显性视神经萎缩;AD:常染色体显性遗传;AR:常染色体隐性遗传;PS:硫代磷酸酯;2’-OMe:2’-氧-甲氧基;2’-MOE:2’-氧-甲氧乙基;/:未知;-:无
2.1 RHO基因相关视网膜色素变性
视网膜色素变性(retinitis pigmentosa,RP)是一种常见的遗传性视网膜变性,有高度的遗传及临床异质性。RHO基因P23H是北美人群常染色体显性RP(autosomal dominant RP,ADRP)的常见致病变异[28]。
RHO基因编码的突变型蛋白P23H会导致视杆细胞丢失和感光细胞变性[29]。保留野生型P23H蛋白表达的同时减少突变型蛋白生成,可以在一定程度上改善ADRP的表型[30]。Murray等[31]研究表明,向RHO基因P23H杂合突变模型小鼠玻璃体内注射靶向2’-MOE-PS gapmer AON能够成功抑制等位基因中突变蛋白的表达并保留野生型蛋白的正常表达,经AON治疗后小鼠感光细胞变性速度得到有效减缓,治疗过程中未观察到毒性及炎症反应。此AON药物已由ProQR公司开发并在2019年获得了美国孤儿药称号,目前正在进行1/2期安全和有效性临床试验。
2.2 NR2E3基因相关RP
1%~2%的常染色体显性遗传RP是由NR2E3基因突变引起。NR2E3与转录因子视锥视杆同源盒(cone-rod homeobox,CRX)及神经视网膜亮氨酸拉链(neural retina leucine zipper,NRL)形成复合体,激活视杆细胞特异性基因表达,抑制视锥细胞特异性基因的表达[21]。NR2E3的突变体p.G56R与CRX结合可破坏复合体的形成,影响光感受器分化和成熟,进而造成RP。
Naessens等[32]分别利用2’-OMe-PS gapmer AON和LNA-PS gapmer AON靶向利用突变NR2E3质粒转染的RPE-1细胞系中产生显性负效应的NR2E3突变体G56R,抑制G56R表达的同时保留了野生型NR2E3的正常表达,从而阻止了光感受器的变性;利用不同的AON策略治疗突变型RPE-1细胞,发现治疗后突变型NR2E3蛋白的表达量降低了1.77%~36.2%,证明AON可以通过等位基因特异性沉默的方式改善由NR2E3基因突变引起的RP,成为一种有潜力的RP治疗手段。
2.3 USH2A基因相关Usher综合征
USH2A基因突变是导致Usher综合征及RP常见原因之一[33]。USH2A蛋白的缺失会导致视网膜光感受器细胞和耳蜗毛细胞静纤毛的变性[34]。
USH2A基因内含子40突变导致剪切异常并产生假外显子40(pseudo exon 40,PE40),导致读码移框且翻译提前停止,产生截短的非功能性USH2A蛋白。Slijkerman等[23]利用2’-OMe-PS-AON靶向患者成纤维细胞中mRNA突变的PE40剪切位点,在跳过PE40后成功恢复正确的USH2AmRNA剪切以及蛋白的表达,实验结果证明AON对于该基因型诱导的Usher综合征有潜在疗效。
另外,在USH2A基因中,13号外显子突变位点(c.2299delG)是欧美国家Usher综合征常见的致病变异,导致阅读框移码并提前出现终止密码子,进而合成截短的非功能性USH2A蛋白。Dulla等[35]通过患者成纤维细胞诱导的诱导性多能干细胞(induced pluripotent stem cells,iPSCs)衍生的光感受器细胞与突变的斑马鱼模型实验证明,利用AON跳跃外显子13后可以合成功能性的USH2A蛋白,并恢复感光细胞和视网膜的功能。
QR-421a药物是经化学修饰的21个核苷酸长度的AON,可以靶向USH2A mRNA并通过13号外显子跳跃的方式产生符合读框规则的转录本,产生略短但功能正常的USH2A蛋白[36]。目前QR-421a正在进行1/2期临床试验。有研究结果显示,8例Usher综合征受试者单眼玻璃体内注射QR-421a后均表现出良好的安全性和耐受性,其中有2例患者显示出良好的治疗效果;应用低剂量和中剂量QR-421a治疗后,外显子13纯合突变和杂合突变的受试者治疗眼全视野ERG暗适应敏感度和视野敏感度均有提升,中剂量组受试者最佳矫正视力有改善,改善作用可持续≥6个月[24]。
2.4 ABCA4基因相关Stargardt病
Stargardt病是一种由于ABCA4基因突变引起的常染色体隐性遗传黄斑营养不良[37]。ABCA4基因编码的三磷酸腺苷结合盒转运子A4蛋白在感光细胞中广泛表达,其基因突变可造成视网膜色素上皮细胞内脂褐质的异常沉积或感光细胞死亡[37-38]。
Albert等[25]研究发现,ABCA4基因中邻近内含子30的突变会造成假外显子PE30插入,并引起ABCA4蛋白截短。在Stargardt病患者iPSCs分化生成的感光前体细胞中,应用不同的2’-OMe-PS AON分别特异性靶向ABCA4mRNA不同突变位点(c.4539+2001G>A;c.4539+2028C>T)可纠正80%的异常剪切,恢复ABCA4蛋白在感光细胞内的正常表达。Sangermano等[39]同样应用2’-OMe-PS AON靶向纠正了ABCA4基因的5个假外显子的插入,全部成功纠正了剪切位点,恢复了ABCA4蛋白的部分功能。以上研究均表明,AON可成为Stargardt病潜在治疗工具。
2.5 CEP290基因相关先天性黑矇
CEP290基因内含子26突变(c.2991+1655A>G)是先天性黑矇10型常见的突变位点之一,会导致假外显子的插入并降低功能性CEP290蛋白的表达,造成光感受器纤毛缺陷,引发严重的视网膜变性[40-41]。
在猴眼玻璃体内注射实验以及先天性黑矇患者iPSCs分化的视网膜类器官模型研究中,2’-OMe-PS AON(QR-110)靶向CEP290 mRNA上c.2991+1655A>G位点进行剪切校正,避免了该突变引导的假外显子插入,恢复了CEP290 mRNA结构功能以及蛋白质的合成,有效提高了纤毛细胞的数量和纤毛长度,进而恢复了光感受器的功能[42]。QR-110的1/2期临床试验已完成,目前正在进行2/3期临床试验,有望成为治疗LCA10的有效候选药物[43]。接受低剂量和中剂量QR-110治疗的受试者最佳矫正视力有改善,全视野ERG暗适应敏感度提高,瞳孔对光反应有所改善;但11例受试者中8例出现晶状体混浊,经白内障手术后恢复原有视力,2例出现轻度黄斑囊样水肿,经局部治疗后消失,2例出现视网膜变薄,在全部剂量注射结束后2个月逐渐恢复正常[26]。
3 AON治疗遗传性眼病的安全性问题
3.1 AON临床治疗的不良反应
AON作为一种特异性较高的小分子靶向药物,具有较高的安全性。目前,AON在眼部治疗引起的非特异性反应主要来自玻璃体内注射过程中产生的局部并发症和炎症反应,如眼内出血、眼内炎症反应和其他注射相关损伤等[44]。AON本身及其化学修饰物有可能引起肝脏和肾脏的不良反应及与蛋白非特异性结合,但是血-眼屏障的存在可以有效减少全身不良反应的发生[45]。在安全性方面,可以进一步通过动物实验判断AON可能引起的各种体内的毒性反应,在临床试验前改善AON的修饰以减少其临床治疗中可能出现的安全性问题。
3.2 AON的药物毒性问题
由欧美国家核酸安全性工作组开展的遗传毒性试验证明多种化学修饰的AON并未表现出遗传毒性。但是由于第3代AON尚未开展遗传毒性试验,并且在理论上存在其与基因组DNA形成3股螺旋导致遗传毒性的可能性,应用前需进一步验证[45]。在使用新型AON的遗传毒性问题上可以通过遗传毒性试验的方式验证,确保AON的高度安全性。
4 小结及展望
随着遗传性眼病发病机制和致病基因研究的不断开展,基因-蛋白-疾病之间的关系不断被揭示,为基因治疗提供了更多新的靶标,同时展示出了基因药物在遗传性眼病临床应用上的巨大潜力。随着递送策略的不断发展,AON的缺陷被不断弥补,逐渐成为基因领域中较为成熟的治疗方式。尤其对于较长基因引起的遗传性眼病,AAV等载体无法实现基因增补,而AON可以弥补这一部分应用的空缺。尽管遗传性眼病的AON在临床试验阶段上刚刚起步,但其安全性及有效性已得到了初步验证。随着基因治疗相关领域的不断探索和AON的进一步优化,AON必将为遗传性眼病的治疗带来更多的可能性。
利益冲突所有作者均声明不存在利益冲突
