葛根素-壳寡糖共无定型制备与体外溶出度评价*
2022-02-17任丽佳唐丽娜
任丽佳,李 杰,唐丽娜△
(1.南京中医药大学江阴附属医院,江苏 无锡214400;2.南京中医药大学附属张家港医院,江苏 张家港 215600)
葛根素是中药葛根中提取的主要有效成分,是一种异黄酮类化合物,临床应用广泛,主要用于治疗心绞痛、高血压、冠状动脉粥样硬化性心脏病等疾病[1−2]。但其水溶性差,口服生物利用度低,临床应用受到限制[3−5]。无定型技术通过破坏药物晶格,可有效改善难溶性药物的溶解度/溶出度。无定型固体分散体(ASD)技术通过添加载体来提高无定型药物的物理稳定性[6−7],载体包括高分子聚合物类和表面活性剂类,但其较强的引湿性均导致物理稳定性差,此外还有与药物混溶性差、胃肠道副作用等问题,且用量较大导致药物服用量大。共无定型技术具有更高的物理稳定性和更好的溶出表现,已成为ASD的替代技术。与单一无定型药物相比,共无定型药物除物理稳定性好外,当配体为活性成分时,还可产生协同治疗、较少副作用等联合治疗效果。壳寡糖也称几丁寡糖,是壳聚糖的水解产物,天然环保,相对分子质量小,水溶性良好,黏度低[8],还具有抗氧化、抗菌、抗炎、抗肿瘤等多种药理活性[9−12]。壳寡糖作为一种生物利用度增强剂,可打开细胞间的紧密连接,提高药物的肠道吸收,增强药物的生物利用度[11−12]。本研究中选择壳寡糖为共无定型的新型药物配体,与葛根素共同制备葛根素−壳寡糖共无定型(PUE−COS CM),采用多种固态表征方法进行分析,并评价其在漏槽和非漏槽2种条件下的体外溶出表现。现报道如下。
1 仪器与试药
1.1 仪器
Waters 2695型高效液相色谱仪(美国Waters公司);Bromanil C18柱(250 mm×4.6 mm,5µm);SPEX 6770型冷冻研磨机(美国SPEX公司);CPA 225D型电子分析天平(南京以马内利仪器设备有限公司,精度为十万分之一);Q2000型示差扫描量热仪(美国TA公司);D/max 2500型X−射线粉末衍射仪(日本Rigaku公司);SNE−4500型扫描电镜仪(日本Hitachi公司);Bruker−TENSOR37型红外光谱仪(德国Bruker Optics公司);HY68/RCY−1型智能溶出试验仪(北京中西远大科技有限公司);LHH−350CFS型综合药品稳定试验箱(上海凯朗仪器设备厂);Milli−Q型超纯水系统(美国Millpore公司);KH−500DE型数控超声清洗仪(昆山超声仪器有限公司,功率为250 W,频率为40 kHz);MX−F型涡旋仪(大龙兴创实验仪器股份<北京>有限公司)。
1.2 试药
葛根素原料药(上海泰坦科技股份有限公司,批号为5214C,纯度大于98%);壳寡糖(浙江金壳药业有限公司,批号为M−KG−1708001,相对分子质量1 000,脱乙酰度大于90%);甲醇(色谱纯,美国Tedia有限公司);氢氧化钠(分析纯,国药集团化学试剂有限公司);葛根素对照品(批号为P111269,纯度大于98%),磷酸二氢钾(分析纯),均购自上海阿拉丁生化科技股份有限公司;水为超纯水。
2 方法与结果
2.1 样品制备
PUE−COS CM:将总质量为1 g的葛根素与壳寡糖[质量比1∶1(m/m)]混匀,采用低温研磨法制备PUE−COS CM。置充满液氮的低温室,以10 Hz的频率研磨5 min,冷却2 min,按此步骤循环,低温研磨30 min。样品置真空干燥箱中干燥24 h,保存于干燥器中,备用。同法制备低温研磨葛根素。
葛根素−壳寡糖物理混合物(PUE−COS PM):取总质量为1 g的葛根素与壳寡糖混合物[质量比1∶1(m/m)],置50 mL离心管中,置涡旋仪上涡旋40 min,使葛根素和壳寡糖充分混匀,即得PUE−COS PM,保存于干燥器中,备用。
2.2 高效液相色谱法测定含量
2.2.1 色谱条件
色谱柱:Bromanil C18柱(250 mm×4.6 mm,5µm);流动相:甲醇−水溶液(51∶49,V/V);流速:1 mL/min;柱温:30℃;检测波长:250 nm;进样量:10µL。
2.2.2 方法学考察
线性关系考察:取葛根素对照品4.825 mg,精密称定,置10 mL容量瓶中,加甲醇适量,超声(功率为250 W,频率为40 kHz)使溶解,定容,振荡摇匀,配制成质量浓度为482.5µg/mL的对照品贮备液。精密吸取500µL,稀 释 成 质 量 浓 度 分 别 为1.88,3.80,7.54,15.08,30.16,60.31,120.63,241.25,482.50 µg/mL的系列对照品溶液,按2.2.1项下色谱条件进样测定。分别以葛根素质量浓度(X,µg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归,得回归方程Y=40 524X−3 145.8(R2=0.999 9,n=9)。结果表明,葛根素质量浓度在1.88~482.50µg/mL范围内与峰面积线性关系良好。
精密度试验:吸取线性关系考察项下质量浓度分别为3.80,60.31,241.25 µg/mL的葛根素对照品溶液,按2.2.1项下色谱条件连续进样测定6次,记录峰面积。结果葛根素峰面积的RSD分别为2.53%,0.98%,1.14%(n=6),表明仪器精密度良好。
稳定性试验:取PUE−COS CM适量,精密称定,置25 mL容量瓶中,加甲醇超声(功率为250 W,频率为40 kHz)使溶解,振荡摇匀,定容,即得葛根素质量浓度为62.16µg/mL的溶液,分别于0,2,4,8,12,24 h时按拟订色谱条件进样测定。结果葛根素峰面积的RSD为1.87%(n=6),表明PUE−COS CM溶液在24 h内稳定性良好。
重复性试验:按稳定性试验项下方法平行制备6份PUE−COS CM供试品溶液,按2.2.1项下色谱条件进样测定。结果葛根素含量的RSD为1.76%(n=6),表明方法重复性良好。
加样回收试验:取6份已知含量的PUE−COS CM适量,精密称定,分别加入等量葛根素对照品,按稳定性试验项下方法制备供试品溶液,按2.2.1项下色谱条件进样测定。结果PUE−COS CM中葛根素的平均加样回收率为96.05%,RSD为2.68%(n=6)。
2.3 PUE-COS CM表征分析
差示扫描量热法:分别取葛根素原料药、壳寡糖、PUE−COS PM、低温研磨葛根素、PUE−COS CM各适量(5~10 mg),置铝坩埚中,置Q2000型示差扫描量热仪中分析,测定条件为,升温速率10℃/min,升温范围50~250℃,氮气流速50 mL/min。结果见图1 A。可见,葛根素原料药在75.4~157.8℃间存在明显失水峰,在191.4~219.2℃间有一尖锐吸热峰,该吸热峰为葛根素原料药的晶体熔化峰(图1 a波);壳寡糖在50.0~145.8℃间存在明显失水峰,在215.2~234.5℃间存在明显吸热峰,该吸热峰由壳寡糖发生降解所致(图1 b波);PUE−COS PM在191.4~234.5℃间存在吸热峰,该峰由葛根素原料药晶体熔化峰与壳寡糖降解峰叠加所致(图1 c波);低温研磨葛根素在191.4~219.2℃间存在葛根素原料药的晶体熔化峰,表明通过低温研磨不能使葛根素原料药晶体完全转变成无定型状态(图1 d波);PUE−COS CM中葛根素的晶体熔化峰完全消失,并存在单一的玻璃化转变温度(95.7℃),表明通过低温研磨成功制备为共无定型系统(图1 e波)。
X射线粉末衍射法:分别取葛根素原料药、壳寡糖、PUE−COS PM、低温研磨葛根素、PUE−COS CM各适量,平铺于载玻片上,置D/max 2500型X−射线粉末衍射仪分析,测定条件为,Cu靶(40 kV,40 mA),扫描范围3°~40°(2θ),扫描速率2°/min,步长0.02°。结果见图1 B。可见,葛根素原料药在6.5°,7.9°,8.7°,11.5°,13.8°,15.9°,16.8°,18.1°,18.9°,19.6°,21.0°,23.4°,26.2°
等处有较强的晶体衍射峰(图1 f波);壳寡糖在3°~40°范围内无晶体衍射峰,表明壳寡糖以无定型形式存在(图1 g波);在PUE−COS PM中,葛根素原料药的晶体衍射峰仍然存在(图1 h波);低温研磨葛根素中,葛根素原料药的晶体衍射峰显著减弱,部分衍射峰消失,但仍存在部分晶体衍射峰,表明低温研磨仅能在一定程度上破坏葛根素原料药的晶格,不能完全使其晶体转变为无定型状态(图1 i波);PUE−COS CM中,葛根素原料药的晶体衍射峰全部消失,表明成功制备了PUE−COS CM(图1 j波)。

a,f,k.葛根素原料药b,g,l.壳寡糖c,h,m.PUE−COS PM d,i,n.低温研磨葛根素e,j,o.PUE−COS CMA.差示扫描量热法B.X射线粉末衍射法C.傅里叶红外光谱法图1表征分析图a,f,k.PUE API b,g,l.COS c,h,m.PUE−COS PM d,i,n.PUE ground by the cryomilling method e,j,o.PUE−COS CMA.DSC B.XRPD C.FTIRFig.1 Phenograms of characterization analysis

A.葛根素原料药B.壳寡糖C.PUE−COS PM D.PUE−COS CM图2扫描电镜图A.PUE API B.COS C.PUE−COS PM D.PUE−COS CMFig.2 SEM images
傅里叶红外光谱法:分别取葛根素原料药、壳寡糖、PUE−COS PM、低温研磨葛根素、PUE−COS CM各适量,采用Bruker−TENSOR37型红外光谱仪分析,测定条件为,扫描波数范围4 000~600 cm−1,分辨率2 cm−1。结果见图1 C。可见,葛根素原料药的主要特征吸收峰为于3 379 cm−1波数处存在-OH的伸缩振动,在1 628 cm−1波数处存在C=O的伸缩振动(图1 k波);壳寡糖的主要特征吸收峰为在1 608 cm−1波数处存在酰胺Ⅰ的C=O伸缩振动(图1 l波);PUE−COS PM中,葛根素和壳寡糖的主要特征吸收峰仍然存在(图1 m波),表明在物理混合物中,葛根素和壳寡糖间未发生相互作用;与葛根素原料药相比,低温研磨葛根素的红外吸收峰未发生明显变化(图1 n波);PUE−COS CM中,葛根素于3 379 cm−1波数处的-OH特征峰消失,壳寡糖在1 608 cm−1波数处的C=O伸缩振动明显减弱,表明葛根素与壳寡糖间发生了潜在的分子间氢键相互作用(图1 o波)。
扫描电镜分析:分别取葛根素原料药、壳寡糖、PUE−COS PM、PUE−COS CM各适量,均匀撒于贴有导电胶带的样品座上,喷金5 min,用镊子取出,置SNE−4500型扫描电镜仪,采用3 000 V的加速电压扫描其表面,并拍照。结果见图2。可见,葛根素原料药具有明显的长片状特征,形状规则(图2 A);壳寡糖为大小不一的规则球形(图2 B);PUE−COS PM中,葛根素原料药的长片状特征和壳寡糖的球形特征仍然存在(图2 C);PUE−COS CM呈颗粒状、不规则块状,且粒径减小,葛根素原料药的长片状特征和壳寡糖的球形特征均完全消失(图2 D)。
2.4 体外溶出试验
漏槽条件下:取葛根素原料药、PUE−COS PM、低温研磨葛根素、PUE−COS CM(相当于6 mg葛根素原料药)各适量,精密称定,按2015年版《中国药典(四部)》附录XC溶出度测定法进行体外溶出试验,溶出介质为磷酸盐缓冲液(pH 6.8),介质体积为900 mL,温度为(37±0.5)℃,转速为50 r/min,分别于2,5,10,20,30,45,60 min时取样3 mL,并随即补足等温度、等量的溶出介质,经0.45 µm微孔滤膜滤过,按2.2.1项下色谱条件进样测定3次,计算各时间点的葛根素累积溶出率。体外溶出曲线见图3 A。可见,在漏槽条件下,葛根素原料药与PUE−COS PM溶出曲线相似;与葛根素原料药相比,低温研磨葛根素的累积溶出率稍提高,主要是由于低温研磨后,葛根素原料药粒径减小,其与溶出介质的接触表面积增大;与葛根素原料药相比,PUE−COS CM的累积溶出率显著提高。5 min时,PUE−COS CM中葛根素的累积溶出率为76.25%,而葛根素原料药仅为35.97%(P<0.01);10 min时,PUE−COS CM中葛根素的累积溶出率为85.26%,而葛根素原料药仅为50.79%(P<0.01)。由于PUE−COS CM自由能更高,且药物粒径减小,导致PUE−COS CM中葛根素的累积溶出率显著提高。
非漏槽条件下:取葛根素原料药、PUE−COS PM、低温研磨葛根素、PUE−COS CM(相当于30 mg葛根素原料药)各适量,精密称定,按2015年版《中国药典(四部)》附录XC溶出度测定法进行体外溶出试验,溶出介质为磷酸盐缓冲液(pH 6.8),介质体积为200 mL,温度为(37±0.5)℃,转速为50 r/min,分别于0.25,0.50,0.75,1.00,1.50,2.00,4.00,6.00 h时各取样2 mL,并随即补足等温度、等量溶出介质,经0.45µm微孔滤膜滤过,用甲醇稀释1倍,按2.2.1项下色谱条件进样测定3次,计算各时间点葛根素的溶出质量浓度。体外溶出曲线见图3 B。可见,在非漏槽条件下,葛根素原料药与PUE−COS PM的体外溶出曲线相似,2 h后趋于稳定;低温研磨葛根素在0.5 h时的溶出质量浓度达最大值(9.25µg/mL),然后下降并趋于平衡,这是由于低温研磨后,葛根素粒径变小,并可能存在部分无定型葛根素,导致溶出质量浓度稍高于葛根素原料药,晶型发生转变后,溶出质量浓度趋向于葛根素原料药平衡状态下的溶出水平;与葛根素原料药和PUE−COS PM比较,PUE−COS CM中葛根素的体外溶出质量浓度较高,且能长时间维持过饱和状态。在1.5 h时,PUE−COS CM中葛根素的溶出质量浓度达最大值(18.31µg/mL),是葛根素原料药(7.65 µg/mL)的2.39倍(P<0.01);在1.5~6.0 h间,PUE−COS CM中葛根素的溶出质量浓度无明显下降,维持长时间的过饱和状态。

A.漏槽条件下B.非漏槽条件下图3葛根素体外溶出曲线A.Under sink condition B.Under non−sink conditionFig.3 Dissolution profiles of PUE in vitro
2.5 稳定性试验
物理稳定性:将PUE−COS CM置LHH−350CFS型综合药品稳定试验箱,在40℃条件下保存6个月。通过X射线粉末衍射法分析,判断PUE−COS CM是否发生晶型转变,以评价PUE−COS CM的物理稳定性。详见图4和图5。由图4可知,PUE−COS CM在40℃条件下保存6个月均未出现葛根素的晶体特征衍射峰,表明PUE−COS CM在6个月内未发生晶型转变,物理稳定性良好。可能由于在PUE−COS CM中,葛根素与壳寡糖分子间发生了氢键相互作用。由图5可知,PUE−COS CM放置6个月的溶出趋势与放置0 d基本一致,且溶出速率良好。这是由于低温研磨不会引起药物的热降解,且葛根素与壳寡糖分子间氢键相互作用较强,导致PUE−COS CM物理稳定性良好。
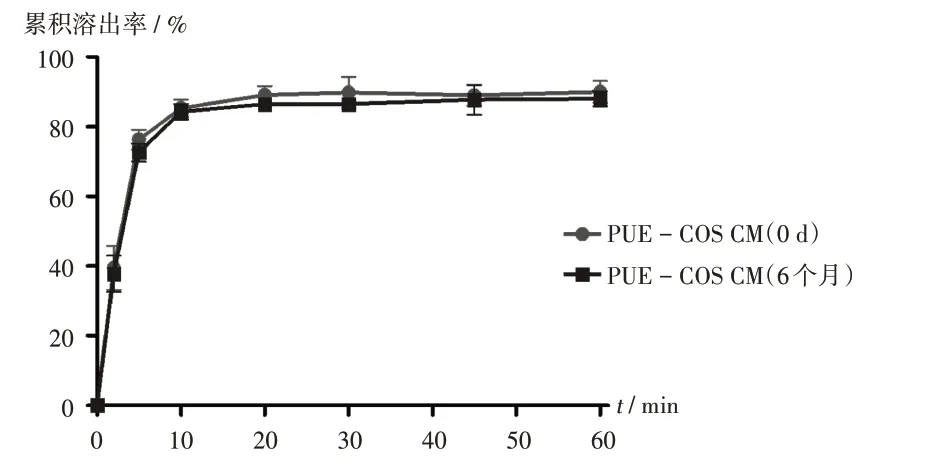
图5 PUE-COS CM 40℃条件下放置0 d和6个月的溶出曲线Fig.5 Dissolution profiles of PUE-COS CM at 40℃for 0 day and six months
化学稳定性:按2.1项下方法制备PUE−COS CM,于40℃条件下放置6个月,按2.2.1项下色谱条件进样测定3次,记录峰面积,并计算含量。结果新鲜制备的PUE−COS CM中葛根素的含量为(0.506±0.005)g/g,40℃条件下放置6个月的PUE−COS CM中葛根素的含量为(0.503±0.008)g/g,表 明PUE−COS CM在40℃条件下放置6个月的化学稳定性良好。
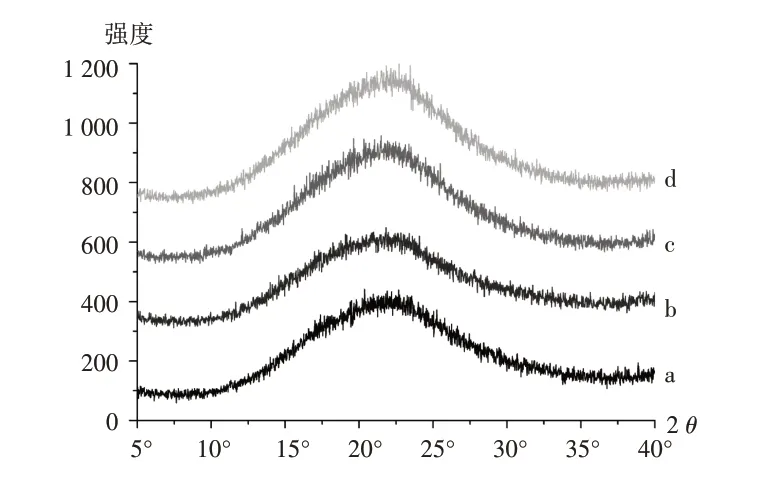
a.0 d b.1个月c.3个月d.6个月图4 PUE-COS CM 40℃条件下放置6个月的X射线粉末衍射图a.0 d b.One month c.Three months d.Six monthsFig.4 XRPD patterns of PUE-COS CM at 40℃for six months
3 讨论
为了提高难溶性药物的溶解度/溶出度,有效方法之一为改变药物晶型,将药物由晶体状态转变为无定型状态[13]。但无定型状态为高能态,热力学不稳定,随着时间的变化易发生重结晶,向稳定的晶型状态转变。目前,为了增加无定型药物的稳定性,通常将药物与高分子聚合物或表面活性剂等载体制备成ASD。当药物与载体混溶性较差时,为了达到药物的有效治疗剂量,一般使用大量的聚合物载体,造成服用量/体积较大。且大量聚合物易吸湿,引起药物转晶,降低ASD的稳定性。此外,使用大量高分子聚合物或表面活性剂还可能引起胃肠道毒性等问题[14−16]。共无定型是一种新型药物递送系统,指由药物和药物或药物和小分子配体组成的二元无定型系统,可有效改善难溶性药物的溶解度/溶出度,且物理稳定性好[17]。共无定型的小分子配体主要包括氨基酸(脯氨酸、精氨酸、苯丙氨酸等)、有机酸(柠檬酸、苹果酸、酒石酸等)等。其中,低分子量糖类(蔗糖、甘露醇、葡萄糖等)作为配体逐渐用于共无定型。壳寡糖为亲水性载体,天然环保,无毒副作用,生物相容性较好,且具有多种药理学活性,展现出作为共无定型优良配体的潜力,已被广泛用于药物制剂开发和生物医学领域,同时可作为吸收增强剂,提高难溶性药物的生物利用度。
本研究中,通过低温研磨成功将葛根素与壳寡糖制备成PUE−COS CM,方法简单易行,并采用多种表征分析技术对PUE−COS CM进行了分析。差示扫描量热和X射线粉末衍射分析结果表明成功制备了PUE−COS CM,这是由于低温研磨破坏了葛根素的晶格,导致其转变为无定型状态;傅里叶红外光谱分析发现,葛根素与壳寡糖分子间发生了潜在的氢键相互作用;扫描电镜观察发现,PUE−COS CM呈不规则块状与颗粒状,未见葛根素原料药的长片状和壳寡糖的球型特征。与葛根素原料药相比,PUE−COS CM在漏槽条件下葛根素的累积溶出速率更快,在非漏槽条件下的溶出质量浓度更高,且可长时间维持过饱和状态。这是由于PUE−COS CM中葛根素和壳寡糖发生了潜在的分子间氢键相互作用,抑制了共无定型中药物的成核和晶体生长[17−18]。此外,在过饱和系统中,药物的浓度高于药物的平衡溶解度,会促进更多游离的药物被人体吸收,且过饱和状态时间维持越长,药物的吸收效果越好[19]。
将葛根素原料药和壳寡糖制备成PUE−COS CM,可有效提高葛根素的体外溶出度,且可长时间维持过饱和状态,提高难溶性药物葛根素的生物利用度,且壳寡糖进一步扩大了共无定型的配体类型,为其他新型药用功能性辅料的开发和应用提供参考,也为解决其他难溶性药物的溶解度/溶出度及生物利用度问题提供新的思路。
