1α,25-二羟基维生素D3通过TLR4信号通路抑制川崎病血清诱导的THP-1细胞炎症因子表达*
2022-02-16李洁滢杨艳娟
周 忠, 田 正, 王 锋, 李洁滢, 胡 琳, 杨艳娟, 焦 蓉
(湖北医药学院附属襄阳市第一人民医院,湖北 襄阳 441000)
川崎病(Kawasaki disease,KD)又称皮肤粘膜淋巴结综合征,其主要临床症状包括发热、结膜充血、草莓舌、皮疹及淋巴结肿大等,最常见的并发症是冠状动脉病变(coronary artery lesion,CAL)[1]。丙种球蛋白联合阿司匹林是KD 治疗的经典方案,早期干预使CAL 的发生率可降至3%~5%。但近年来不完全KD 病例数量呈上升趋势,使得KD 诊断及治疗更加困难[2-3]。目前其发病机制尚不明确,有研究表明Toll 样受体(Toll-like receptor,TLR)途径参与了KD的发病机制[4]。TLR4途径是巨噬细胞抵御外界病原的重要通路,但过度活化的TLR4途径是造成炎症损伤的重要环节[5]。有关TLR4 途径在KD 发病中发挥的作用尚无定论。
维生素D3是体内调节钙磷代谢的重要类固醇激素。但很早以前人们便发现通过光照可改善结核患者的症状,随着后来研究的深入人们逐渐认识到维生素D3是体内重要的免疫调节剂。其可调节单核细胞、巨噬细胞、树突状细胞和淋巴细胞等发挥抗炎的生物学效应[6]。当病原体入侵后,刺激巨噬细胞体内的维生素D 在25-羟基维生素D3[25-hydroxyvita‑min D3,25-(OH)D3]-1α-羟化酶(CYP27B1)的羟化作用产生1α,25- 二羟基维生素D3[1α,25-dihy‑droxyvitamin D3,1,25-(OH)2D3],与核内维生素D 受体(vitamin D receptor,VDR)结合,可导致TLR2 及TLR4 炎症通路下调,白细胞介素12(interleukin-12,IL-12)产生减少,保护性炎症因子IL-10 上调[6-8]。Stagi 等[9]发现,急性期KD 患儿体内25-(OH)D3的水平显著下降,推测维生素D3水平的降低与KD患儿体内炎症反应的发生发展有关,但目前相关研究报道较少。因此,本研究拟通过观察1,25-(OH)2D3对TLR4 途径的影响,探讨维生素D3在KD 中发挥抗炎作用的机制。
材料和方法
1 对象与材料
1.1 研究对象选取2019 年9 月~2021 年5 月本院住院治疗KD 患儿73 例。纳入标准:(1)符合日本KD 研究会的诊断标准,年龄5 岁以下;(2)73 例患儿入院时均处于发病的10 d以内。排除标准:(1)入院前3 月已使用丙种球蛋白、肾上腺皮质激素及抗血小板等治疗,近半年服用维生素D治疗;(2)近1月内有感染性疾病病史或合并其他免疫性疾病;(3)不同意、不配合研究者。另外选取我院儿保科正常体检儿童及急性上呼吸道感染(acute upper respiratory tract infection,AURI)患儿各30 例作为正常组和感染组(受试对象近半年均未服用维生素D)。所有受试对象均处于丙种球蛋白治疗前,采集静脉血3 mL,并于当天分离血清,冻存于−80 ℃冰箱。利用简单随机抽样的方法从73 例KD 血清中随机选取40 例,与30 例感染组及30 例正常体检儿童血清比较,应用25-(OH)D3试剂盒(化学发光法)检测各组25-(OH)D3水平。另外33 例KD 血清吸取相同体积后进行混合,并在56 ℃水浴锅加热30 min后用于细胞实验,保证实验中血清的同质化处理。所有受试对象家属均知情同意,项目已通过医院学术伦理委员会批准。
1.2 实验材料及主要试剂人急性单核细胞白血病细胞THP-1(货号CC-Y1519)购自EK-Bioscience;鼠抗人β-actin 抗体(Proteintech,66009-1-Ig);兔抗人磷酸化NF-κB P65 抗体(Wanleibio,WL02169);兔抗人TLR4 抗体(Absin,abs132000);反转录试剂PrimeScript RT Master Mix 和实时定量试剂SYBR Premix®ExTaqTM(TliRNaseH Plus)均购自TaKaRa;羊抗兔549 Ⅱ抗(货号A23220)购于Abbkine。
2 方法
2.1 细胞培养THP-1细胞培养于RPMI-1640完全培养液(10% 胎牛血清、1% 青霉素/链霉素)中,在37 ℃、5% CO2及饱和湿度条件下培养。传代时控制细胞密度在(2~4)×108/L,并在细胞生长至(8~10)×108/L 时进行传代,约每3 d 传代1 次。取处于对数生长期的细胞进行实验。
2.2 THP-1细胞的分组和处理将THP-1细胞分别以1×109/L 和5×108/L 的密度培养于6 孔板和24 孔板中,以100 nmol/L佛波酯(phorbol 12-myristate 13-ace‑tate,PMA)诱导THP-1 细胞24 h,使其向巨噬细胞转化。在光学显微镜下观察细胞的形态变化,证实诱导成功。利用15% 同质化处理后的KD 血清刺激诱导后THP-1 细胞24 h 作为KD 固有免疫模型。用不同浓度1,25-(OH)2D3(1、10 和100 nmol/L)预保护48 h 后,应用15% KD 血清刺激THP-1 细胞24 h 作为维生素D 干预组。实验处理及实验分组如下:15% 正常血清组、15% KD 血清组及15% KD 血清+1,25-(OH)2D3(1、10 和100 nmol/L)组。本研究中使用的1,25-(OH)2D3浓度(1、10 和100 nmol/L)与正常维生素D 剂量给药后健康人群血浆中的浓度一致[10]。分别提取细胞中RNA、总蛋白和细胞培养上清液,采用RT-qPCR 检测TLR4 通路及相关炎症因子mRNA 的表达;采用Western blot 检测全细胞裂解物TLR4 和磷酸化NF-κB P65(p-P65)的蛋白水平;采用ELISA法检测细胞培养上清液中肿瘤坏死因子α(tumor ne‑crosis factor-α,TNF-α)的水平;采用免疫荧光技术检测细胞TLR4 蛋白表达水平。每项实验至少进行3次重复实验。
2.3 RT-qPCR将密度为1×109/L 的THP-1 细胞接种于6 孔板中,取对数增殖期的细胞进行干预,按照如上方式进行PMA 诱导,以无血清不同浓度(1、10和100 nmol/L)的1,25-(OH)2D3进行预保护20 h,最后按照上述分组加入正常血清及KD 血清刺激24 h。利用Trizol 试剂提取6 孔板中不同处理细胞中总RNA,取1 μg 总RNA 按逆转录试剂盒说明逆转录成cDNA,取2 μL 为模板进行PCR。总反应体系为20 μL。反应条件为:95 ℃预变性3 min;95 ℃变性20 s,58 ℃复性30 s,72 ℃延伸45 s,40个循环。以β-actin作为内参照,用2−ΔΔCt法计算TLR4、IL-1β 和TNF-α mRNA的相对表达量。引物序列见表1。

表1 引物序列Table 1.Sequences of the primers
2.4 Western blot培养于6孔板的THP-1细胞处理方式同上,1,25-(OH)2D3预保护48 h 后,加用血清刺激24 h,弃去培养液,PBS 冲洗2 遍后,采用蛋白裂解液在冰上裂解细胞15 min,裂解后4 ℃、13 500×g离心15 min 得到蛋白上清液。利用BCA 蛋白浓度测定,测定蛋白浓度。蛋白变性后,每孔上样量为30 μg,预先制备10%聚丙烯酰胺凝胶电泳,采用聚偏二氟乙烯膜进行转膜(250 mA,150 min)。在5% 脱脂牛奶封闭2 h 后,将膜与抗TLR4 和抗p-P65 Ⅰ抗(1∶1 000)4 ℃孵育过夜。在TBST 洗涤后,用辣根过氧化酶标记的Ⅱ抗孵育2 h。Western blot 条带采用ChemiDocTMXRS+ 成像系统(Bio-Rad)成像,利用Image Lab 凝胶分析软件分析,以目的蛋白与内参照条带累积吸光度之比作为蛋白水平的相对指标。
2.5 免疫荧光染色为进一步了解单核细胞TLR4的表达情况,采用免疫荧光检测TLR4的表达。将密度为5×108/L 的THP-1 细胞接种于多聚赖氨酸包被的载玻片上,实验分组:15% 正常血清组、15%KD 血清组、15%KD 血清+100 nmol/L 1,25-(OH)2D3组和15% 正常血清+100 nmol/L 1,25-(OH)2D3组。1,25-(OH)2D3预保护48 h 后,加用血清刺激24 h。用PBS洗涤3次,用4%多聚甲醛固定30 min,然后在室温下用专用封闭液封闭和透膜,与抗TLR4 Ⅰ抗(1∶300)孵育过夜。PBS清洗细胞3次后,与DyLight 549标记的山羊抗兔Ⅱ抗(1∶1 000)室温孵育1 h,细胞核用Hoechst 33342 染色20 min。选择合适的滤光片,在荧光显微镜下采集图像。
2.6 ELISA 法将密度为5×108/L 的THP-1 细胞接种于12孔板中,1,25-(OH)2D3预保护48 h,收集血清刺激24 h后各组培养上清液测定其中炎症因子的表达情况,人TNF- α ELISA 检测试剂盒购自Multi‑Sciences(货号70-EK182HS),操作步骤严格按说明书进行。
3 统计学处理
采用SPSS 26.0 软件处理数据,计量资料以均数±标准差(Mean±SD)表示,两组间比较采用独立样本t检验和单因素方差分析。P<0.05 认为差异有统计学意义。
结 果
1 KD组与感染组人口学资料及实验室检查的比较
收集73 例KD 患儿及30 例AURI 患儿基本情况及实验室数据进行比较。2 组年龄比较无统计学差异;与感染组相比,KD 组患儿白细胞数、中性粒细胞数、血小板数、丙氨酸转氨酶及C-反应蛋白均显著升高(P<0.01),而体内白蛋白水平较感染组呈显著下降(P<0.01);2 组间血红蛋白及血钠水平差异无统计学意义(P>0.05),见表2。
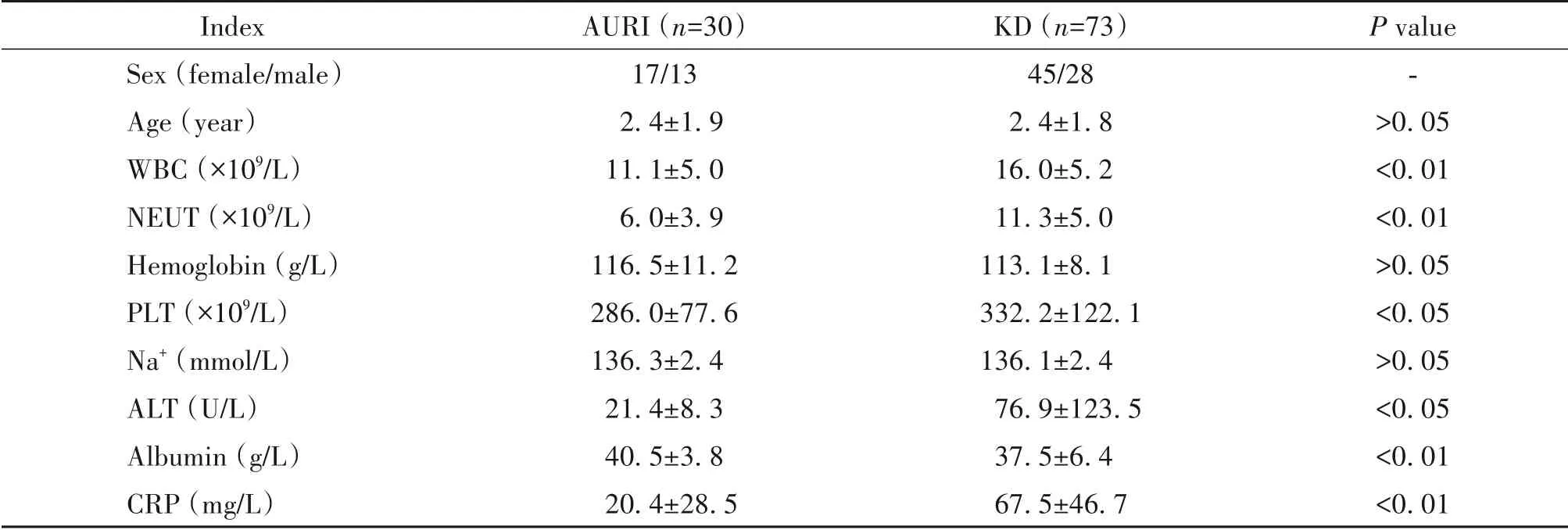
表2 KD组和感染组临床指标的特点Table 2.Clinic indexes in Kawasaki disease(KD)group and acute upper respiratory tract infection(AURI)group(Mean±SD)
2 KD 组、感染组与正常组血清中25-(OH)D3水平的比较
应用试剂盒检测40 例KD 组、30 例感染组及30例正常组血清中25-(OH)D3水平,分别为(21.86±7.41)、(33.39±8.96)和(35.04±5.14)μg/L;KD 组25-(OH)D3水平较感染组显著下降(P<0.01),与正常组比较也得到同样的结果(P<0.01),感染组和正常组血清中25-(OH)D3水平差异无统计学意义(P>0.05),见图1。
3 血清25-(OH)D3水平预测KD的ROC曲线
ROC 曲线显示,血清25-(OH)D3水平预测KD 的曲线下面积为0.922(95% CI:0.864~0.980),与正常组相比差异有统计学意义(P<0.01);将25-(OH)D3取截断值为27.55 μg/L 时,预测KD 的灵敏度为93.3%,特异度为75.0%,见图2。
4 应用PMA诱导THP-1细胞转化为巨噬细胞
应用100 nmol/L PMA 对悬浮生长的人单核细胞诱导24 h,诱导单核细胞向巨噬细胞转化。如图3所示,分别在不同倍镜下观察THP-1 细胞形态,A~C 为PMA 诱导前细胞形态,D~F 为PMA 诱导后细胞形态。光学显微镜下可见单核细胞贴壁生长,由圆形变为椭圆形、不规则形,伸出伪足,证实单核细胞已被成功诱导为巨噬细胞。

Figure 1.Serum 25-(OH)D3 level in 40 patients with Kawasaki disease(KD),30 healthy controls,and 30 acute up‑per respiratory tract infection (AURI) controls.Mean±SD.**P<0.01 vs healthy and AURI control groups.图1 三组血清中25-(OH)D3水平
5 1,25-(OH)2D3显著抑制血清诱导的THP-1 巨噬细胞促炎因子的表达
RT-qPCR 结果显示,与正常血清诱导组相比,KD 血清诱导组可显著促进巨噬细胞IL-1β 和TNF-α的mRNA 表达水平显著升高(P<0.05 或P<0.01);以不同浓度的1,25-(OH)2D3预保护20 h后可显著降低KD 血清诱导的巨噬细胞IL-1β 和TNF-α 的mRNA 表达(P<0.05),见图4B、C。ELISA 结果显示,与正常血清组相比,KD 血清刺激巨噬细胞可使TNF-α 的分泌水平显著升高(P<0.05);而1,25-(OH)2D3预保护可显著抑制TNF-α的分泌(P<0.05),见图4D。

Figure 2.The ROC curve of serum 25-(OH)D3 level predicting Kawasaki disease(KD).There were 40 patients with KD,and 30 healthy controls.图2 血清25-(OH)D3水平预测川崎病的ROC曲线
6 1 ,25-(OH)2D3显著抑制巨噬细胞TLR4和p-P65的活化
Western blot 结果显示,与正常血清相比,KD 患儿血清可刺激巨噬细胞TLR4 和p-P65 蛋白表达(P<0.05);1,25-(OH)2D3在浓度为100 nmol/L 时可显著抑制KD 血清诱导的TLR4 和p-P65 蛋白表达(P<0.05),见图5。此外,1,25-(OH)2D3可降低血清诱导的TLR4 mRNA 表达水平(P<0.01),见图4A。同样的,TLR4免疫荧光染色结果与Western blot得到相同的结论,如图6 所示,与正常血清组相比,KD 血清诱导巨噬细胞表达TLR4荧光强度更强(P<0.05);应用100 nmol/L 的1,25-(OH)2D3预保护48 h 后,TLR4 荧光强度显著下降(P<0.05)。

Figure 3.Phorbol 12-myristate 13-acetate(PMA)induced the transformation of THP-1 cells into macrophages.Human THP-1 mono‑cytes were induced by 100 nmol/L PMA for 24 h.A,B and C:morphological changes of THP-1 cells before PMA induc‑tion;D,E and F:morphological changes of THP-1 cells after PMA induction.Scale bar=200 μm in A and D;scale bar=100 μm in B and E ;scale bar=20 μm in C and F.图3 佛波酯诱导THP-1细胞转化为巨噬细胞

Figure 4.Effect of 1,25-(OH)2D3 on serum-induced expression of pro-inflammatory factors in THP-1 cells.A,B and C:the mRNA expression levels of TLR4,IL-1β and TNF-α in KD serum-treated THP-1 cells after pretreatment with 1,25-(OH)2D3;D:the secretion of TNF-α in the cell culture supernatant detected by ELISA.Mean±SD. n=3.#P<0.05,##P<0.01 vs control(Con)group;*P<0.05,**P<0.01 vs KD group.图4 1,25-(OH)2D3对血清诱导的THP-1细胞TLR4及促炎因子mRNA表达的影响
讨 论
我们的研究观察到维生素D3对KD 起着至关重要的保护作用,这种保护作用是通过抑制巨噬细胞TLR4 信号通路激活来实现的,这一以前未引起我们重视的类固醇激素为KD 的诊断、评估和治疗提供了新的线索。
自从1967 年川崎富作医生报道了50 例KD 以来,50 多年过去了。目前其病因机制尚不清楚,感染、遗传、免疫及环境因素均影响着疾病的发生和发展[11-12]。日本流行病学调查显示KD 发病有明显的季节性,主要好发于冬春季节[13]。而冬春季节的北半球平均日照时间缩短,皮肤接受紫外线照射时间也缩短。而在紫外线照射下体内7-脱氢胆固醇才可最终转化为维生素D3最终形式1,25-(OH)2D3,作为体内的活性成分在多种生理过程中发挥作用[14]。Stagi等[9]的研究提出低水平的25-(OH)D3与KD冠脉并发症的发生具有相关性。陈小红等[15]的研究也证实25-(OH)D3与CAL 发生有关,但其无法区分KD 和健康儿童。而本研究结果表明KD 患儿体内25-(OH)D3显著低于普通上呼吸道感染及正常儿童。因此我们认为KD 免疫炎症反应的发生可能与体内保护性活性成分维生素D3水平下降有关。
目前尽管对于不完全KD 的诊断提出不少的预测评分系统,但不同国家间评分系统的可靠性有待考究。更加可靠的危险评分指标的加入,可能增加评分的可靠性。关于25-(OH)D3水平作为KD 独立的危险因素及预测指标的研究较少。因此,我们进一步评估了血清25-(OH)D3水平作为预测KD 指标的可靠性,结果显示ROC 曲线下面积为0.922,灵敏度为93.3%,特异度为75.0%,诊断的截断点为27.55 μg/L,其作为KD 预测指标有较好的灵敏度和特异度。
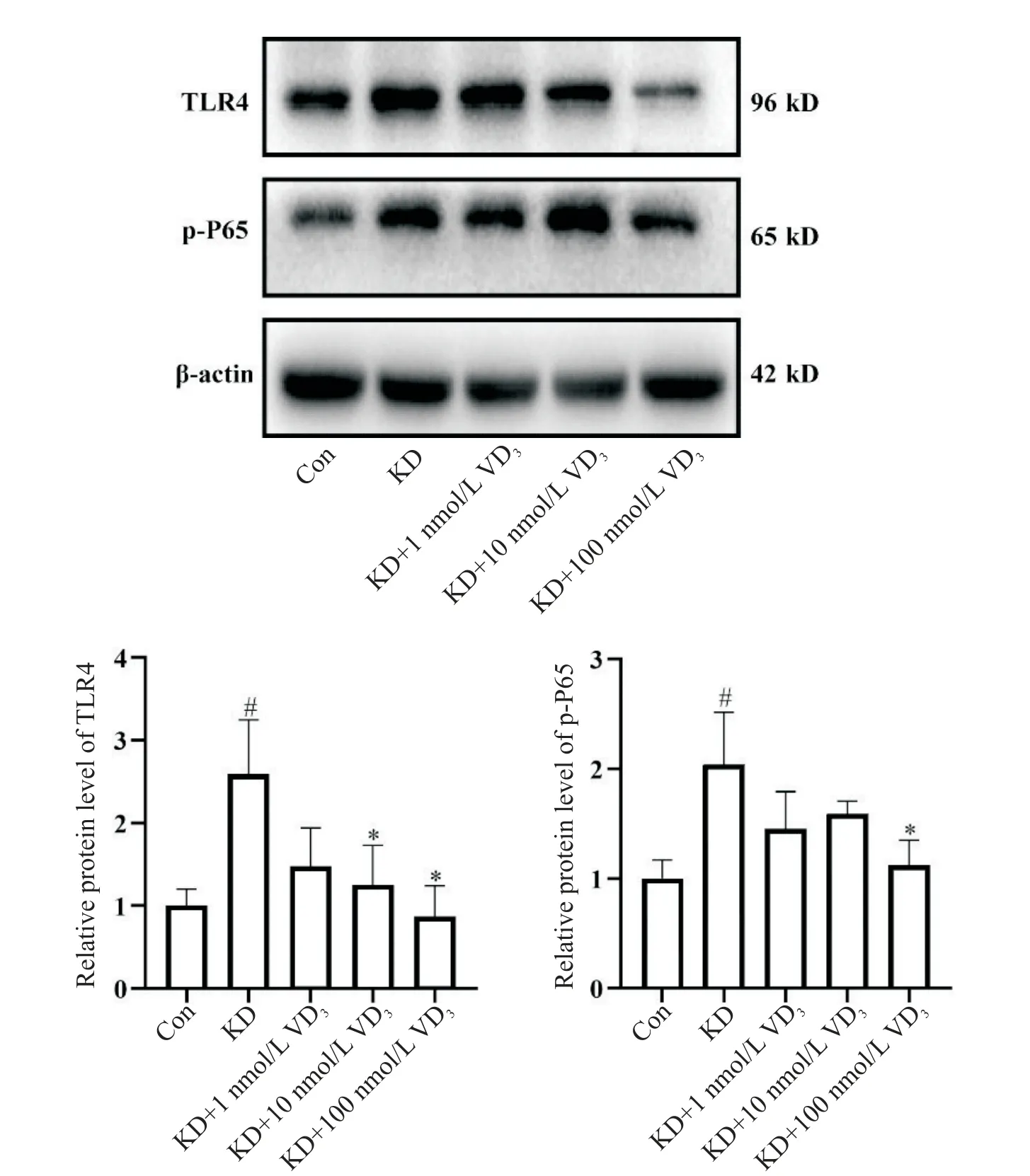
Figure 5.1,25-(OH)2D3 significantly inhibited the activation of TLR4 and NF-κB in macrophages.The protein levels of TLR4 and p-NF-κB P65 were detected by Western blot.Mean±SD. n=3.#P<0.05 vs control(Con)group;*P<0.05 vs KD group.图5 1,25-(OH)2D3对THP-1细胞TLR4和NF-κB活化的影响
巨噬细胞TLR4途径在KD 的发病机制和进程中扮演着重要的作用。基于单细胞测序的结果,KD 患儿较正常儿童体内外周血单个核细胞中单核细胞占比显著升高,关键炎症因子IL-1β 和TNF-α 的差异也来自单核细胞[16]。正因如此我们选用单核巨噬细胞系作为干预对象,以期望从上游环节发现KD 的发病机理。G-菌产生的脂多糖(LPS)、G+菌产生的磷壁酸酯质及内源性配体等病原相关分子模式(pathogenassociated molecular patterns,PAMP)通过与巨噬细胞Toll 样受体结合,进而通过NF-κB 活化,NF-κB 核转位进入细胞核与相应启动子结合,诱导前炎症因子及细胞表面共刺激分子转录[17]。王国兵等[4,18]对KD 患儿外周血巨噬细胞的研究显示,急性期KD 患儿TLR4、MyD88 及MD-2 mRNA 显著增高。此外,他们的研究表明急性期KD 存在TLR4-MyD88 非依赖性途径异常活化。他们提出KD 体内PAPMs 可能触发巨噬细胞TLR4,但并未得到证实。但Rosenkranz等[19]在应用干酪乳杆菌构建KD 小鼠模型的研究中表明,KD 小鼠冠脉炎症反应的发生依赖于TLR2 信号通路而不是TLR4通路。目前关于TLR4途径是否参与KD 炎症反应发生尚无定论,本研究中应用KD患儿血清诱导人单核巨噬细胞,从基因和蛋白水平表明急性期KD 患儿血清中存在某种PAPM 激活巨噬细胞TLR4 炎症信号通路,并活化NF-κB 途径上调IL-1β 和TNF-α 的mRNA 表达,显著提高了关键性炎症因子TNF-α 的含量。进一步证实巨噬细胞TLR4活化可能是KD免疫功能紊乱的始动因素之一。
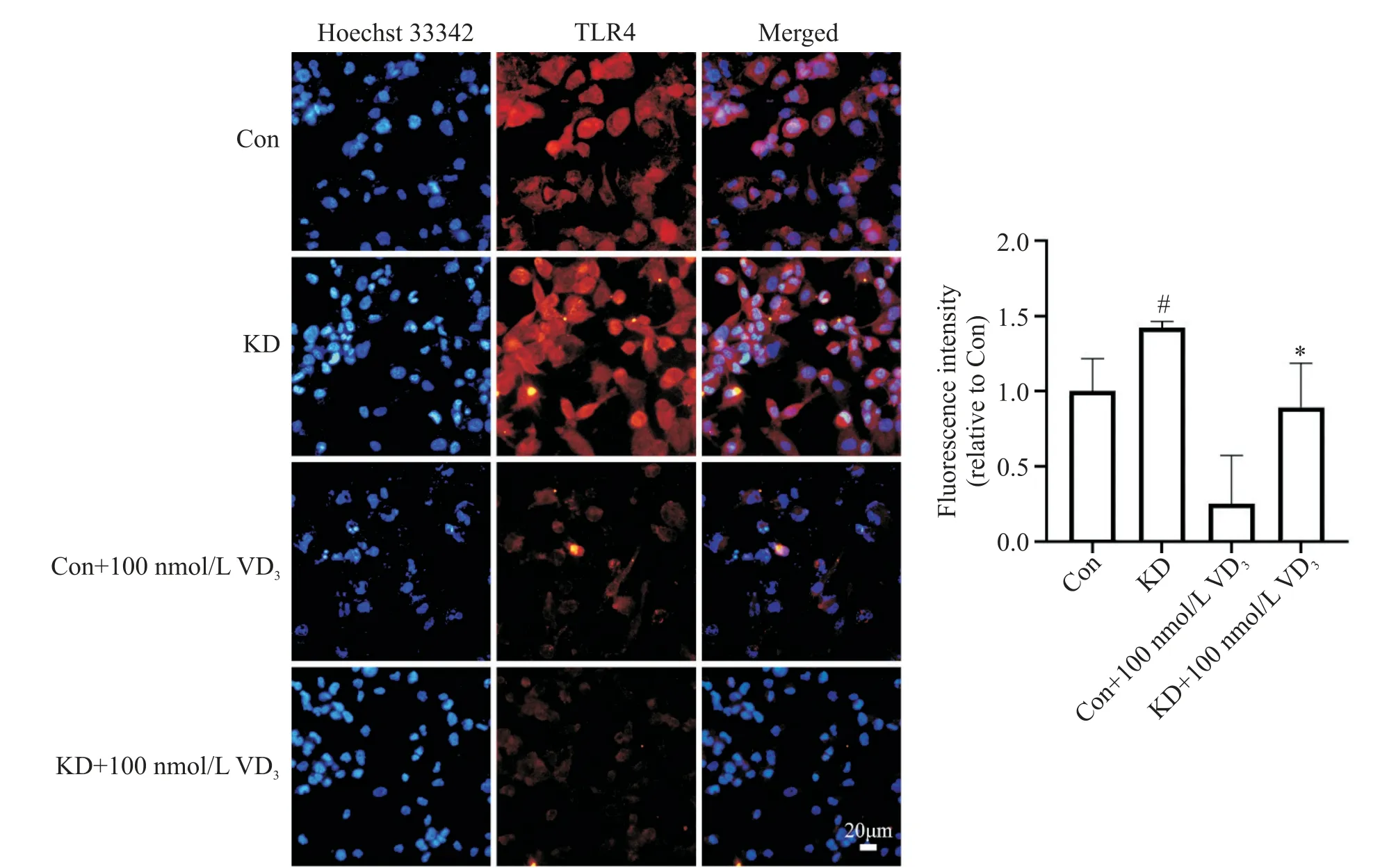
Figure 6.1,25-(OH)2D3 significantly inhibited the activation of TLR4 in macrophages.The expression of TLR4 was detected by im‑monofluorescence staining.Mean±SD. n=3.#P<0.05 vs control(Con)group;*P<0.05 vs KD group.图6 1,25-(OH)2D3显著抑制巨噬细胞TLR4的活化
我们观察到KD 患儿体内的25-(OH)D3水平呈显著下降,同时基于维生素D 在免疫调节中的重要作用,我们提出额外提高维生素D 的水平是否能在KD 患儿体内发挥保护作用。Suzuki 等[20]的研究显示,1,25-(OH)2D3通过抑制NF-κB 途径,从而减轻TNF-α 诱导人冠脉内皮细胞的炎症反应,提示维生素D 可在冠脉炎症中发挥抗炎作用。而维生素D 是否能通过巨噬细胞来影响KD 炎症反应尚未见报道。我们的研究表明,1,25-(OH)2D3可显著抑制KD 血清诱导的巨噬细胞TLR4 途径的活化,进而抑制NF-κB信号转导,最终抑制关键性炎性因子的产生和分泌。此外,有文献报道1,25-(OH)2D3通过活化P53 通路并抑制ERK1/2 通路调节T 细胞的增殖,抑制T 细胞在KD 患儿体内过度增殖[21]。综上所述,证明维生素D 不仅可能从上游免疫细胞环节抑制KD 炎症风暴的发生,还可抑制炎症因子导致的冠脉内皮细胞损伤。基于糖皮质激素及乌司他丁等在儿童应用的副作用问题以及生物制剂(英夫利昔单抗、依那西普和阿那白滞素等)的经济负担,维生素D 可能是一种更安全更经济的治疗手段。但关于维生素D 是否能作为KD 辅助治疗方案,尚缺乏多中心临床研究确定其疗效,值得进一步研究。
基于上述结果,我们推测在环境因素的影响下,KD 易感儿童受到外界病原刺激,由于体内维生素D产生不足,巨噬细胞TLR4 炎症途径过度活化,从而激活NF-κB 途径启动炎症因子的转录和翻译。而补充维生素D 通过下调巨噬细胞TLR4 和NF-κB 信号通路,从而抑制KD 患儿炎症级联效应。但目前有关维生素D 在KD 的发病机理中的作用上尚未完全阐明,补充维生素D 对KD 及冠脉损伤是否有预防及改善作用,仍有待进一步探讨。
