肝细胞癌免疫相关lncRNA预后风险模型的建立与评估
2022-02-15彭双谭英征杨秋红易来
彭双,谭英征,杨秋红,易来
(中南大学湘雅医学院附属株洲医院1.感染内科2.血液科,湖南株洲412007)
在全球范围内, 肝细胞癌(hepatocellular carcinoma,HCC)是排名第6 位的恶性肿瘤,2018年全球约有841 080 例新发病例和781 631 例死亡病例[1]。根据美国疾病控制与预防中心的数据,2009—2016年之间,HCC 的病死率上升了25%[2]。尽管目前对HCC 的治疗有手术治疗、肝移植、射频消融、栓塞治疗、靶向治疗和放化疗等多学科综合治疗,但HCC 的病死率和复发率仍呈上升的趋势[3-4]。因此在临床中迫切需要采取新的干预措施来改善HCC 患者的预后。HCC 通常起源于慢性肝损伤,尤其是乙型病毒性肝炎[5]。对HCC 患者的预后评估将有助于制定更好的治疗策略。常规临床实践中已采用了多种分期系统,例如巴塞罗那临床肝癌系统[6]。但是,目前尚未建立基于分子生物标志物的临床分层系统来识别高危HCC 患者。
长非编码RNA(long noncoding RNA,lncRNA)是一类长度超过200 个核苷酸的非编码RNA。越来越多的研究表明,lncRNA 在调节广泛的生物学过程中起着重要作用,包括基因表达,细胞生长,分化和染色体分裂[7]。在各种类型的癌症中都检测到异常表达的lncRNA,并且某些lncRNA 表现出致癌或抑制肿瘤的作用,这表明了其可能作为肿瘤潜在的生物标志物和治疗靶点[8-9]。并且研究发现与mRNA 和微小RNA(miRNA)相比,lncRNA 是卓越的肿瘤生物标志物[10-11]。目前,尽管对lncRNA的生物学功能的报道相对较少,但越来越多的研究表明lncRNA 可以为多种类型的肿瘤提供重要的预后价值[12-15]。随着新一代测序技术的应用和普及,大量有关肿瘤的高通量测序数据使得利用生物信息学预测肿瘤中lncRNA 功能成为可能。
本研究中,通过利用TCGA 数据库中公开获取的HCC 转录组测序数据和临床资料,对HCC 免疫相关lncRNA 进行了全面的分析,建立HCC 预后免疫相关lncRNA 预后风险模型并进行评估,这将对HCC 管理和治疗策略的制定有很大帮助。
1 材料与方法
1.1 材料
从癌症基因组图谱(The Cancer Genome Atlas,TCGA)(https://cancergenome.nih.gov)下载HCC 转录组数据和临床资料,下载截止日期到2020年10月9日,临床资料包括:性别、年龄、肿瘤分级,肿瘤病理分期,TNM 分期,生存时间和生存状态,排除生存时间<30 d、肿瘤病理分级缺失、TNM 分期缺失以及生存信息不详的病例[16],共下载到376 例HCC 转录组数据和相应的临床资料,根据排除标准,共纳入225 例HCC 样本。
1.2 获取HCC lncRNA表达矩阵
从TCGA 数据库中下载HCC 相对应的metadata和manifest,通过Perl 语言脚本提取HCC 原始counts数据表达矩阵, 从Ensembl 网站(https://asia.ensembl.org/index.html)下载Homo-sapiens.GRCh38.95.chr.gtf.gz 文件,通过Perl 语言脚本对原始counts 数据进行ID 转换得到gene symbol 表达矩阵,再使用Perl 语言脚本提取HCC lncRNA 表达矩阵。
1.3 HCC免疫相关lncRNA的筛选
从GSEA 网站(https://www.gsea-msigdb.org/gsea/index.jsp) 获取免疫相关基因集列表(Immune system process M13664,Immune response M19817),从225 例HCC 转录组数据中获得的lncRNA 和免疫相关基因集数据,利用R 语言的“limma”包以共表达分析计算lncRNA 与免疫相关基因的相关性,以相关系数Cor>0.6 和P<0.001 筛选HCC 免疫相关lncRNA[17]。
1.4 HCC预后免疫相关lncRNA的筛选及预后风险模型构建
利用Perl 语言脚本将HCC 生存时间和免疫相关lncRNA 表达矩阵进行合并,使用“survival”包进行单因素Cox 回归分析评估免疫相关lncRNA 的预后价值,以P<0.05 筛选预后相关lncRNA,纳入多因素Cox 回归分析进一步筛选,根据赤池信息量准则(Akaike information criterion,AIC) 确定最佳lncRNA 建立预后风险模型,计算每例样本的风险评分(risk score),计算公式为:所纳入的预后免疫相关lncRNA 的基因表达量乘以多因素Cox 回归系数之和。根据中位风险评分值将HCC 患者分为低风险组和高风险组。
1.5 统计学处理
利用Kaplan-Meier 生存分析法比较低风险组和高风险组患者的生存差异,并绘制预后风险模型的生存曲线,同时采用ROC 曲线下面积AUC 值评估预后风险模型的特异度和敏感度。通过单因素和多因素Cox 回归分析评估HCC 患者风险评分和临床病理信息与总生存期的关系,并绘制森林图,筛选HCC 预后的独立危险因素,P<0.05 为差异有统计学意义。
2 结 果
2.1 病例特征
本研究共纳入225 例HCC 病例,平均年龄57.03(17~80)岁,性别、年龄、肿瘤分级,肿瘤病理分期和TNM 分期(表1)。
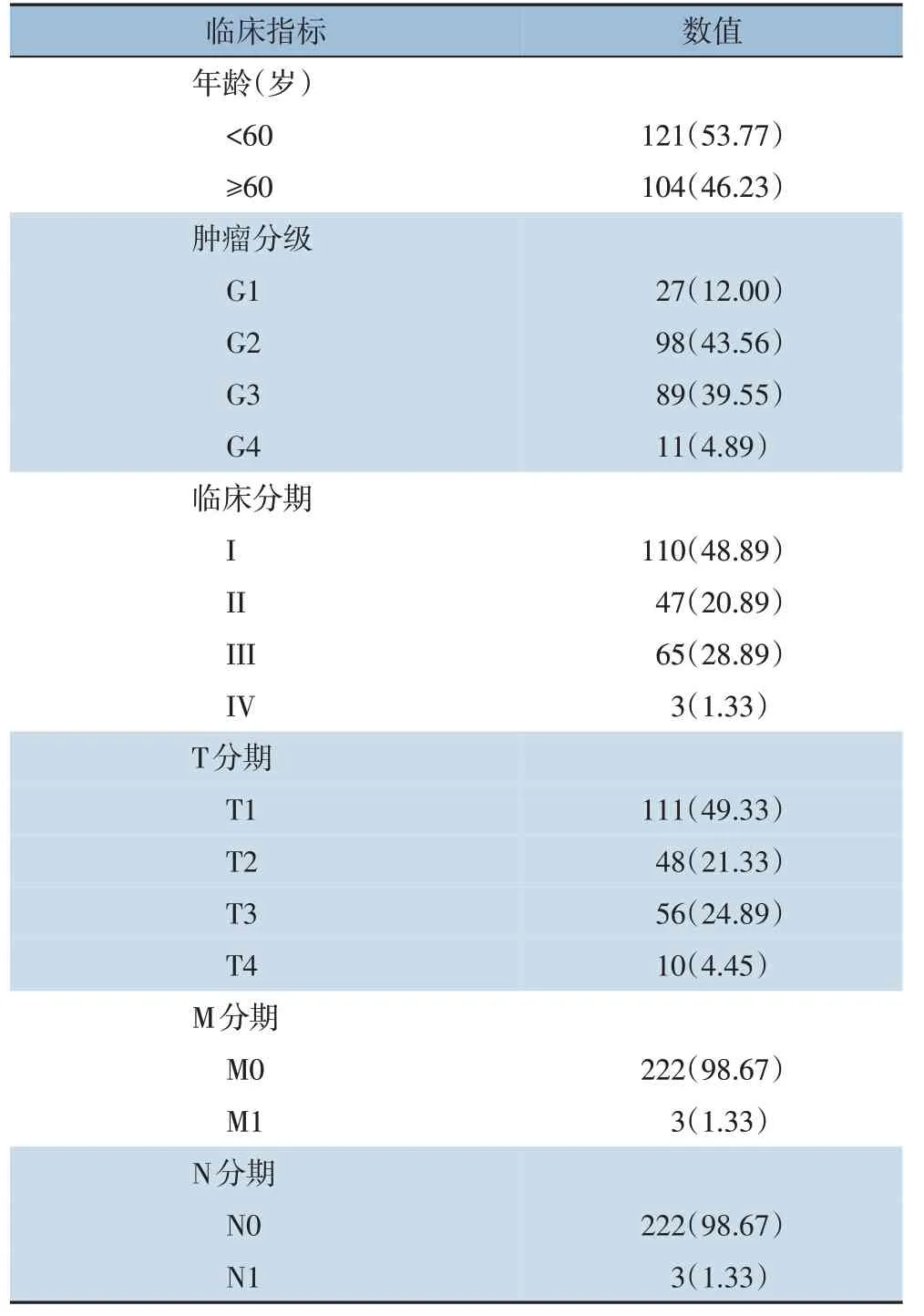
表1 225例HCC患者临床病理特征[n(%)]Table 1 Clinicopathologic characteristics of 225 patients with HCC[n(%)]
2.2 HCC预后免疫相关lncRNA 的筛选
从225 例HCC 转录组数据中获取lncRNA 和免疫相关基因集数据,利用R 语言的“limma”包以共表达分析计算lncRNA 与免疫相关基因的相关性,以相 关系 数Cor>0.6 和P<0.001 提 取HCC 免 疫相 关lncRNA 143 个。使用“survival”包进行单因素Cox回归分析筛选出预后免疫相关lncRNA 17 个(均P<0.05)(表2)。
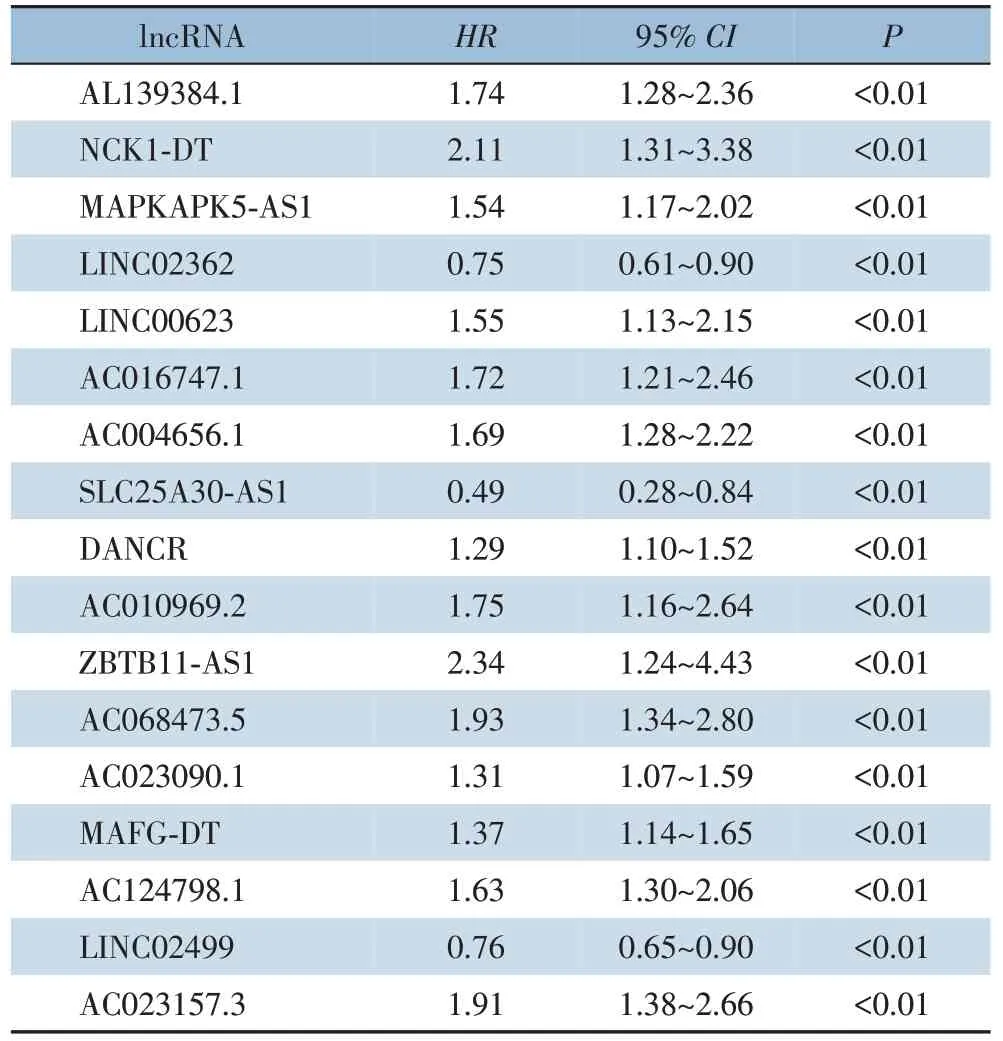
表2 单因素Cox回归分析筛选HCC预后免疫相关lncRNATable 2 HCC Prognostic immune-related lncRNAs screened by univariateCox regression analysis
2.3 免疫相关的lncRNA 预后风险模型的建立与评估
对于预后免疫相关lncRNA 进一步纳入多因素Cox 回归分析进行筛选,根据最优AIC 值纳入8 个lncRNA 构建预后风险模型(表3),计算每例样本的风险评分(risk score)(图1),根据中位风险评分值将HCC 患者分为低风险组和高风险组(图1)。Kaplan-Meier 生存分析法比较两组患者的生存差异,结果显示,高风险组患者生存率明显低于低风险组患者(P<0.05),生存曲线显示随着风险评分越高,患者生存率越低(图2)。随后,通过绘制预后风险模型ROC 曲线评价模型的特异度和敏感度,曲线下面积AUC 值为0.774,说明该模型具有良好的效能(图3)。

表3 多因素Cox 回归分析筛选免疫相关lncRNA 构建HCC预后风险模型Table 3 Construction of HCC prognostic risk model using the immune-related lncRNAs screened by multivariate Cox regression analysis
2.4 预后风险模型的临床应用
进一步探索预后风险模型在HCC 患者预后中的应用价值,通过单因素和多因素Cox 回归分析评估HCC 患者风险评分和临床病理信息与总生存期的关系,结果显示,风险评分为HCC 患者预后的独立预测因子,该预后风险模型可以有效预测患者的生存预后(HR=1.608,95%CI=1.351~1.913,P<0.001)(图4)。
3 讨 论
HCC 是一种高度恶性肿瘤,被列为肿瘤病死率的第三大原因。尽管在HCC 的诊断和治疗方面已取得了巨大进展,但HCC 患者总生存率的提高仍进展缓慢[6,18]。为了寻找新的生物标志物来改善HCC 患者的预后,越来越多学者和研究人员开始关注lncRNA,发现lncRNA 与肿瘤发生发展有关,在HCC 中lncRNA 出现异常表达,并且与肿瘤的生长和患者的预后不良密切相关[19-20]。
过去,研究主要集中在mRNA 和miRNA,对于大多数lncRNA 的功能了解甚少。随着高通量测序数据的快速发展,使得研究学者预测某些转录本的生物学功能的得以实现。对lncRNA 功能的预测也成为研究的热点,同时促进了lncRNA 在肿瘤中的探索。最近有研究[21-22]报道了基于自噬相关lncRNA 的预测模型在HCC 中的应用,并且发现模型对HCC 具有预后判断价值。同时也有学者[23]报道了基于多基因lncRNA 模型在HCC 中的研究,发现lncRNA 模型可良好地预测HCC 患者的生存。此外研究也发现基于免疫相关基因的预测模型也可以有效预测HCC 患者的预后[24]。但是目前尚无免疫相关lncRNA 预后风险模型在HCC 中的研究报道,在本研究中,对TCGA 数据库中HCC 患者的lncRNA 表达谱和相关临床信息进行了全面分析。提取HCC 免疫相关lncRNA 143 个。使用“survival”包进行单因素Cox 回归分析筛选出免疫预后显著相关lncRNA 17 个,Cox 多因素回归分析进一步确定8 个lncRNA 构建预后风险模型,并且根据风险模型计算每位患者的风险评分,将患者分为低风险组和高风险组,高风险组患者生存率显著低于低风险组患者,用于预测HCC 患者生存率的风险评分模型的ROC 曲线下面积AUC 达到0.75 以上,表明在生存预测方面具有良好的特异度和敏感度。此外,单变量和多变量Cox 回归分析表明,风险评分为HCC 患者预后的独立预测因子,证明该预后风险模型可用于预测HCC 患者的预后。在8 个lncRNA 中 AL139384.1、 DANCR、 AC124798.1 和AC023157.3为危险基因, MAPKAPK5-AS1、LINC02362、SLC25A30-AS1 和LINC02499为保护基因。研究[21,25-26]发现DANCR 在HCC 中出现高表达与肿瘤发生显著相关,并且与HCC 预后显著相关。此外,在结直肠癌中发现MAPKAPK5-AS1 在肿瘤组织中显著高表达,敲除MAPKAPK5-AS1 可以显著抑制结直肠癌细胞增殖并引起细胞凋亡。MAPKAPK5-AS1 可作为结直肠癌新型的预后生物标志物[27-28]。同样有研究[29-30]报道LINC02499 为HCC的保护基因,可能是潜在的HCC 诊断和预后生物标志物,并可能在HCC 的发生发展中发挥抑癌作用。而另外5 个lncRNA 目前尚无相关研究报道,仍有待进一步研究。
综上所述,基于TCGA 数据HCC lncRNA 表达谱的全面分析,最终确定了8 个预后免疫相关lncRNA 构建的预后风险模型,可以有效预测HCC患者预后和危险分层的分子标志物,可作为其独立的预后生物标志物。但本研究存在一定的局限性,包括缺乏多中心数据库的验证,目前8 个lncRNA 在HCC 中分子作用机制和相关信号通路尚不清楚等,仍需进一步研究。
利益冲突:所有作者均声明不存在利益冲突。
