Menkes病1例家系报道并中国临床表型和基因型文献复习
2022-02-14潘瑞英
潘瑞英
(中国人民解放军联勤保障部队第九二四医院儿科,广西 桂林 541002)
Menkes病是一种X-连锁隐性遗传病,由ATP7A基因突变导致铜转运障碍所引起的多系统疾病。典型的Menkes病临床特征为毛发卷曲、稀疏,癫痫,神经退行性变和结缔组织异常,通常3岁前死亡[1-3]。随着国内对本病认识的提高和基因检测技术的快速发展,病例报告数增多,本研究报道1例Menkes病患儿的临床表型和基因型,并对国内报道的文献进行复习,总结我国Menkes病的临床表型和基因型,特别是基因检测方法的选择,以提高我国基层医院对该病的诊断能力,避免漏诊,同时通过加强遗传咨询,产前诊断,提高优生优育。
1 资料与方法
1.1 临床资料 患儿,男,1岁8个月,主因间断抽搐3个月余,咳嗽1个月,再发抽搐1 d于2019年3月6日入本院。患儿系第9胎第5产,其母因胚胎停育引产1次,产检发现胎儿畸形引产1次,自行要求引产2次,患儿3个姐姐分别15、14、6岁,均体健,其一兄生长发育落后,重度营养不良,11个月大时因先天性斜疝并嵌顿未及时手术死亡。该患儿系足月顺产,出生体质量2.95 kg,出生后喂养困难,生长发育落后,现仍不能抬头,不能坐、爬、翻身、走等,生后满1岁时因先天性巨结肠、肠梗阻、肺炎、脑积水、运动发育迟缓至广西医科大学第一附属医院住院,行结肠造瘘手术及对症治疗,好转出院。于1岁5个月时因重症肺炎、呼吸衰竭、横结肠造瘘术后、生长发育迟缓再次至广西医科大学第一附属医院住院治疗,期间出现频发抽搐,予对症处理好转出院。住院期间外送武汉康圣达医学检验行全外显子基因检测,出院时结果尚未回报。本次因发热咳嗽及再发抽搐入本院。入院查体:体质量8 kg,经皮血氧90%,精神反应差,抬头不稳,皮肤白,高腭弓,头发稀疏、卷曲,短而粗硬(见图1),呼吸急促,可见吸气性三凹征,双肺满布湿啰音,四肢肌张力高,姿势异常,双上肢背伸,双手外旋,拇指内收,双下肢伸直,双足尖足。
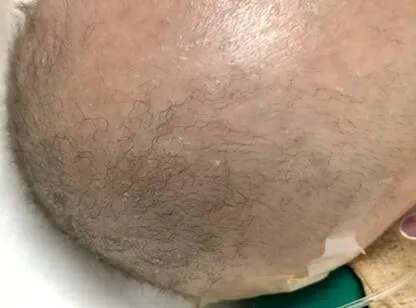
图1 患儿头发表型Figure1 Hair phenotype of the Menkeschild
1.2 一般检查 脑电图、头颅MRI、血清铜、血浆铜蓝蛋白、血尿遗传代谢疾病筛查、血尿生化检查、染色体核型分析、MLPA(多重连接探针扩增技术)。
1.3 ATP7A基因检测 采集患儿及家系成员外周血,应用EDTA全血(乙二胺四乙酸抗凝血)行安捷伦外显子芯片捕获+高通量测序。并对发现的ATP7A基因突变位点用Sanger测序法验证(武汉康圣达医学检验所)。
1.4 文献复习 在万方数据库中检索关键词为“Menkes”,检索时间为建库至2019年3月31日。检索文献共28篇,综述2篇[4-5],Menkes病影像学病例报告3篇[6-8],Menkes病护理报道2篇[9-10]以上病例均包含在各医院的临床病例报道中,不重复统计。其中病例报道21篇中,同一医院同一时间,证实为相同患儿的不重复统计。文献中女性患儿尸解诊断1例,未行基因学检测,未计入[11]。综上,复习文献[12-26]诊断Menkes患儿共40例。
2 结果
2.1 临床特点 患儿男性,足月出生,体质量适于胎龄儿,生后发育落后,喂养困难,特殊外貌(肤色白皙、高腭弓、毛发卷曲稀疏短粗硬),癫痫发作,合并先天性巨结肠肠道畸形。
2.2 一般检查结果 既往广西医科大学第一附属医院检测:血清铜6μmol/L(正常值12.6~29.9μmol/L),血浆铜蓝蛋白80 mg/L(正常值180~450 mg/L);脑电图:散在尖波、棘慢综合波;头颅MRI:脑积水;血串联质谱和尿气相色谱分析未见异常;入院前桂林医学院第一附属医院:染色体核型分析为46,XY;MLPA未见染色体微重复或微缺失。本次入院胸片:双肺炎症,血氨88.5μmol/L,血气分析提示低氧血症及高乳酸血症,乳酸波动于2.8~5.2 mmol/L,肝功能、肾功能、心肌酶、凝血功能无明显异常。
2.3 基因检测结果ATP7A基因外显子11位点发生c.2455G>T,无义突变,染色体位置chrx-77270207,其突变来自于其母亲,其父无突变,患儿大姐、三姐为杂合突变,其二姐无突变;一代测序验证图,见图2。

图2 患儿家系一代测序结果Figure2 First-generation sequencing results of the Menkeschild'sfamily
2.4 患儿家系图谱 患儿家系图谱,见图3。

图3 患儿家系图谱Figure3 Family map of the Menkes
2.5 治疗结果 丙戊酸钠控制癫痫,持续气道正压通气(CPAP)呼吸支持,头孢哌酮钠舒巴坦钠控制感染及呼吸道管理,住院9 d,抽搐获得控制,肺炎明显好转,家属要求自动出院。
2.6 文献复习临床表型 40例患儿全部为男性,运动发育落后100%,头发卷曲稀疏100%,其次高发临床表型为皮肤白皙和惊厥发作,均为92.50%(37/40),另3例患儿单纯因运动发育落后就诊,就诊时尚未出现惊厥。惊厥发生年龄3 d~7个月,平均(3.25±1.43)个月。患儿肌张力表现不一,但主要为肌张力降低62.5%(25/40),肌张力增高7.50%(3/40),肌张力正常30.00%(12/40)。随访到16例具体死亡年龄为50 d~4岁,平均(15.0±12.1)个月。其他伴随异常多为喂养困难,高腭弓,皮肤松弛,面颊虚胖,伴发其他畸形为先天性斜疝,漏斗胸,膀胱憩室,听力异常等。辅助检查:所有惊厥患儿脑电图均提示典型癫痫波形,头颅MRI非特异性改变,表现为脑白质发育不良、脑白质髓鞘化延迟、脑萎缩、胼胝体变薄、脑积水、硬膜下积液等,部分行MRA提示脑血管迂曲变形。检验:全部病例均血清铜降低和或铜蓝蛋白降低。文献检索临床表型,见表1。

表1 Menkes病临床表型Table1 Clinical phenotypes of Menkes disease
2.7 文献复习基因型及基因检测方法文献复习40例患儿行基因检测共25例,具体基因型及基因检测方法,见表2。

表2 Menkes病基因型及基因检测方法Table 2 Menkes disease genotype and gene detection method
3 讨论
1962年,Menkes等[27]首次描述5例Menkes病男婴。SMITH人类先天性畸形图谱[28]指出,该病常见临床表型,①生长发育落后;②中枢神经系统:大脑退行性变,1~2个月时出现严重的进行性神经缺陷伴肌张力增高,癫痫发作,颅内出血,喂养困难;③面部:缺乏表情,面颊虚胖;④头发:稀疏,短而粗硬,着色浅,放大镜显示毛发卷曲和部分断裂;⑤皮肤:偶尔变厚,相对较干,出生时着色不均;⑥骨骼病变:干骺端变宽,肋骨和股骨明显,易折断;⑦眼睛:近视,斜眼;齿龈增厚和乳牙出牙延迟;胃息肉伴胃肠出血,幽门狭窄,食管裂孔疝;膀胱憩室等。通常该病婴儿早期即出现进行性恶化,3年内死亡。结合本例患儿及复习国内病例报道,其临床表型表现为生长发育落后、惊厥发作、头发卷曲稀疏、皮肤白、肌张力减低或肌张力增高,最常见的面部畸形为高腭弓,其他并发畸形包括先天性斜疝、肾盂扩张、膀胱憩室、巨结肠等,尚无幽门狭窄、食管裂孔疝的报道,有视网膜发育不良的报道,无近视报道,皮肤的临床表型除颜色白皙外,变厚干燥着色不均等均未见报道。辅助检查特征性指标,血铜、铜蓝蛋白降低。基因型:本病为X-连锁隐性遗传病,致病基因为ATP7A(GenBank NM_000052)突变,该基因定位于Xq13.3,长度为140×103包含23个外显子。该突变导致编码铜转运ATP酶活性降低甚至丧失,导致机体铜离子缺乏,继而导致各种铜依赖酶功能障碍。目前已知300多种ATP7A基因突变,突变类型包括大片段缺失、错义突变、无义突变、剪接位点突变等。国内报道[12,14-16,19-20,22-24]基因型涵盖上述所有突变类型,未发现明显的热点突变。突变来自母亲携带者占64%(16/25),其他为新生突变。基因检测方法国内文献[12,14-16,19-20,22,24]报道多采用PCR直接测序,全外显子二代测序,或癫痫panel的二代测序。临床表型高度怀疑本病但上述检测阴性情况下,加做MLPA,该技术结合探针杂交和PCR技术,针对待测DNA靶序列进行定性和半定量的分析方法,可用于检测大片段缺失和重复。但该方法不适合检测未知的点突变类型。直接PCR测序+MLPA,或全外显子二代测序+MLPA共同应用可提高致病性突变的检出率。基因检测的目的除明确诊断外,在遗传咨询和优生优育方面发挥重要作用。用于携带者母亲再次怀孕的产前诊断,及子女中女性携带者婚育时的遗传咨询。
综上所述,Menkes病是一种X连锁的隐性遗传病,致病突变来自携带者母亲,或新发突变。临床上针对婴儿期起病的癫痫发作,伴明显的神经运动发育落后,及典型的毛发改变,需警惕该病发生的可能。完善血清铜和血清铜蓝蛋白的检测及结合特殊MRA影像学检查可临床诊断,基因二代测序技术加MLPA技术完成基因诊断。该病目前无有效的治疗方法,预后极其不良,组胺酸铜治疗仍处于试验阶段,国内仅报道1例患儿自6个月大时定期皮下注射组胺铜,患儿仅血铜基本正常,但仍存在智力发育落后和频发癫痫发作[15]。基因诊断的目的不仅是先证患儿的明确诊断,最重要的是Menkes病家庭可通过基因诊断获益。
