Mitochondrial hepatopathy: Anticipated difficulties in management of fatty acid oxidation defects and urea cycle defects
2022-02-12AathiraRavindranathMoinakSenSarma
Aathira Ravindranath, Moinak Sen Sarma
Aathira Ravindranath, Division of Pediatric Gastroenterology, Institute of Gastrointestinal Sciences, Apollo BGS Hospitals, Mysore 570023, Karnataka, India
Moinak Sen Sarma, Pediatric Gastroenterology, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow 226014, Uttar Pradesh, India
Abstract Fatty acid oxidation defects (FAOD) and urea cycle defects (UCD) are among the most common metabolic liver diseases.Management of these disorders is dotted with challenges as the strategies differ based on the type and severity of the defect.In those with FAOD the cornerstone of management is avoiding hypoglycemia which in turn prevents the triggering of fatty acid oxidation.In this review, we discuss the role of carnitine supplementation, dietary interventions, newer therapies like triheptanoin, long-term treatment and approach to positive newborn screening.In UCD the general goal is to avoid excessive protein intake and indigenous protein breakdown.However, one size does not fit all and striking the right balance between avoiding hyperammonemia and preventing deficiencies of essential nutrients is a formidable task.Practical issues during the acute presentation including differential diagnosis of hyperammonemia, dietary dilemmas, the role of liver transplantation, management of the asymptomatic individual and monitoring are described in detail.A multi-disciplinary team consisting of hepatologists, metabolic specialists and dieticians is required for optimum management and improvement in quality of life for these patients.
Key Words: Mitochondrial hepatopathy; Metabolic liver disease; Liver transplantation; Hyperammonemia; Hypoglycemia; Carnitine
INTRODUCTION
Mitochondria occupy a unique place in the metabolic milieu which orchestrates complex physiological processes in response to nutrient signals.Beta oxidation of fatty acids and the urea cycle are among the myriad metabolic functions in which mitochondria play a central role.While beta-oxidation involves energy extraction from fats, the urea cycle converts excess nitrogen into urea and both of these processes encompass several steps with multiple enzymes.Defects in any of the steps would result in the accumulation of the intermediate metabolites and deficiency of the downstream products.Appropriate management mandates an in-depth understanding of the biochemistry and the clinical implications of defects in various steps.
FATTY ACID OXIDATION DEFECTS
Epidemiology
Fatty acid oxidation defects (FAOD) are rare metabolic disorders and the true prevalence is difficult to determine as newborn screening cannot detect all the cases especially the milder ones.FAODs were detected in 1:6560 samples by newborn screening with tandem mass spectrometry in a study from Singapore[1].The incidence of FAOD was determined to be 1:9300 from newborn screening results that included 5256999 samples from Australia, Germany and the United States of America.The incidence varied from 1:3300 in Turkey to 1:217000 in Taiwan[2].Thus, the incidence of FAOD depends on the geographical region and ethnic background.In a large French study, Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency was found to be the most common (25%) followed by long-chain hydroxyacyl CoA dehydrogenase (LCHAD) deficiency (22%).Very long-chain acyl CoA dehydrogenase (VLCAD) deficiency and carnitine shuttle defects accounted for 19% each[3].The reported incidence of defects in MCAD is 1:4000 to 1:15000, VLCAD is 1:85000, LCHAD/trifunctional protein (TFP) is 1:250000 and carnitine shuttle defects is 1:750000[4].Clinical manifestations will present before 2 years of age in 85% of cases and 30% of them will present in the neonatal period although FAOD can present for the first time even in early adulthood[4].
Beta oxidation in brief
Fats are highly concentrated sources of energy that are the ideal reservoir in all the animals as they are non-polar, occupy less space and can be oxidized multiple times to yield more energy compared to carbohydrates and proteins[5].Though fats undergo alpha and gamma oxidation, beta-oxidation is the most important process by which the energy trapped in fats gets converted to adenosine triphosphate (ATP).Triglycerides are first oxidized by lipoprotein lipase to fatty acids which are then shuttled across the plasma membrane to the cytosol by fatty acid transport proteins.These proteins have acyl CoA synthetase activity which converts fatty acids into acyl CoA after translocation.The carnitine shuttle (Figure 1) aids in transporting acyl CoA esters across the mitochondrial membrane.Carnitine palmitoyl transferase 1 (CPT-1) first converts acyl CoA to acyl carnitine that is shuttled through the mitochondrial membrane by carnitine acyl carnitine translocase.At the inner mitochondrial membrane carnitine palmitoyl transferase 2 (CPT-2) retransforms acyl carnitine to acyl CoA and free carnitine.Acyl CoA undergoes beta-oxidation (Figure 2) in which by a series of reactions in each cycle two-carbon units are released as acetyl CoA and also nicotinamide adenine dinucleotide (NADH) and Flavin adenine dinucleotide (FADH2) are produced[5].Acetyl CoA enters the citric acid cycle, NADH and FADH2 enter the electron transport chain.Beta oxidation has four major steps: First, acyl CoA dehydrogenase forms enoyl-CoA on which hydratase will act to generate hydroxyl-acyl CoA.Hydroxy-acyl CoA is dehydrogenated to keto-acyl CoA.Lastly, thiolase will cleave the keto-acyl CoA to acetyl CoA and acyl CoA (with two carbons less).There are three forms of acyl CoA dehydrogenases in humans which are chain length specific-VLCAD, MCAD and Short chain acyl CoA dehydrogenase (SCAD).Short and long chain enoyl CoA hydratases as well as short chain hydroxyl acyl CoA dehydrogenase (SCHAD) and LCHAD are present for metabolizing chain length-specific fatty acids.Similarly, two different thiolases are also present long chain and medium chain[6].The mitochondrial trifunctional protein has hydratase, hydroxyl acyl CoA dehydrogenase and thiolase activity.Defects in fatty acid oxidation can have effects on multiple organ systems including the liver, muscle, heart, brain and kidneys.Thus, presentations are variable and making a quick and accurate diagnosis is a real challenge.
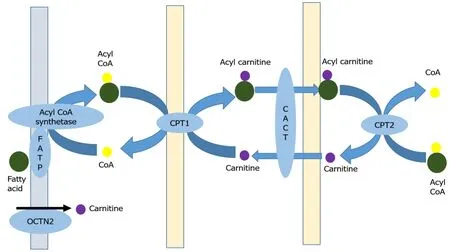
Figure 1 Carnitine shuttle.
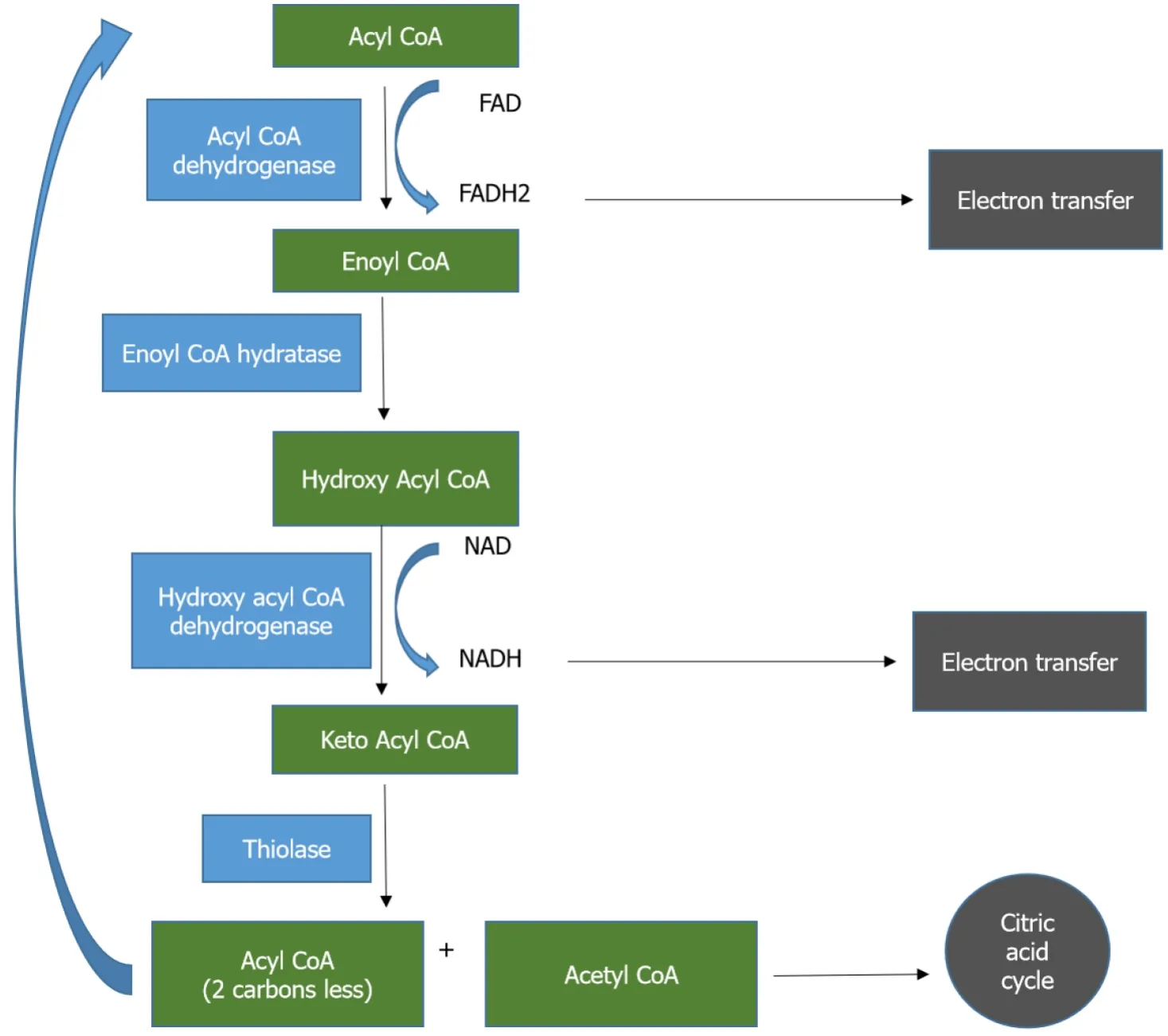
Figure 2 Beta-oxidation of fatty acids.
There are many dilemmas while managing FAODs
With varying clinical presentations in different age groups and multi-system involvement, work-up for FAOD needs to be considered based on detailed knowledge of the manifestations.
Interpreting fatty acid metabolite results based on the severity of the clinical situation and identifying the confounders that can affect the values.
Appropriate dietary management in different age groups.
Asymptomatic FAODs detected on newborn screening mandates a different approach based on the type of FAOD.
Status of the newer therapeutic agents in the overall management.
When not to supplement carnitine and medium-chain triglycerides (MCT).
Challenges in diagnosing FAOD
The common underlying factor in all FAODs is metabolic stress due to fasting, infection or muscular stress.During normalcy, these conditions will stimulate fatty acid oxidation for the ketone body and ATP generation.Defects in any of the enzymes involved in beta-oxidation will result in excess acylcarnitine intermediates accumulation and diversion to omega oxidation that would generate toxic dicarboxylic acids[7].The probability of detecting abnormal fatty acid metabolites would be maximum during a metabolic crisis and finding a normal profile during a mild episode does not rule out FAOD.Hypoglycemia occurs due to increased glucose utilization and absent gluconeogenesis due to deficiency of acetyl CoA (generated during beta-oxidation) and it is typically hypoketotic.Acetyl CoA is also required for the generation of Nacetyl glutamate (NAG) that activates carbamoyl phosphate synthetase in the urea cycle.As a consequence, conversion of ammonia to urea is impaired which results in hyperammonemia.Since oxidative phosphorylation is impaired in FAOD, pyruvate oxidation takes place leading to lactic acidosis that is more pronounced in long-chain disorders.Acyl CoA accumulation causes lipid peroxidation and steatohepatitis[8].
Neonatal period:The predominant presentation in the first month of life is cardiac arrhythmias, conduction defects, hypotonia, lethargy, coma in the background of facial dysmorphism, hypoketotic hypoglycemia, hyperammonemia, lactic acidosis.Renal dysplasia and brain malformations may also accompany.Sudden death due to arrhythmias is seen in about 7% of cases with a neonatal presentation[9].Hepatomegaly (93%), increase in transaminases (68%), cholestasis (36%) and liver failure (25%) are seen in the first two months of presentation[3].
Infancy:Hepatic involvement predominates during infancy when children present with Reye-like syndrome, hepatomegaly, hypoketotic hypoglycemia and steatosis.Hepatic failure may be encountered in 10% of infants and rarely cholestasis.Hyperammonemia, lactic acidosis, mild elevation of serum transaminases can be seen.Cardiomyopathy and skeletal myopathy can be seen in about 50%.
Childhood and adolescence:Apart from typical hepatic manifestations, muscular manifestations are frequently seen in older children with episodic muscular pains, rhabdomyolysis and myoglobinuria.Retinitis pigmentosa and peripheral neuropathy are specifically seen in those with LCHAD defects.
Predominant symptoms and biochemical findings in individual FAODs are given in Table 1.Though dividing the predominant manifestation as hepatic, neurological, muscular, cardiac and renal is easy to understand, a given case will have various permutations and combinations of these presentations.Moreover, most of the episodes may be triggered by an infection and differentiating between severe sepsis and FAOD also is a formidable task.The other metabolic diseases that closely resemble FAODs are urea cycle defects (UCD), mitochondrial hepatopathies, organic acidemias andgluconeogenic disorders.Characteristic hypoketotic hypoglycemia would be the first step to differentiate FAODs from the other disorders.Blood and urine samples for fatty acid metabolite estimation ought to be drawn before carnitine supplementation otherwise the results would be erroneous.Targeted gene panel analysis in children presenting with hypoglycemia can reliably diagnose inborn errors of metabolism including FAOD[10].

Table 1 Characteristic findings in clinically significant Fatty acid oxidation defects
Management dilemmas
Since there are no large-scale trials on the management of individual FAODs, treatment recommendations are based on consensus by experts based on retrospective studies.The rarity of the individual FAODs precludes the conduction of randomized controlled trials.The cornerstone of treatment in all FAODs remains the prevention of hypoglycemia especially during times of metabolic stress so that beta-oxidation is not triggered.Recommendations and issues in the management of the discrete disorders are given below.
Carnitine transport defect
Plasma carnitine levels are very low (< 5 μmol/L) and renal losses are high.L-carnitine supplementation at a dose of 100-400 mg/kg/d in three divided doses remains the mainstay of treatment[11].Targeting the normal plasma carnitine level of 20 to 50 μmol/L is desirable by titrating the dose, however, large doses of carnitine can cause diarrhea and abdominal discomfort.Regular measurement of plasma carnitine levels during well periods should be done to determine the appropriate oral dosage.Even if plasma carnitine levels normalize with supplementation, intracellular carnitine levels in myocytes do not increase by more than 10% of normal due to the absence of active transport.The accurate plasma carnitine level that can ensure adequate passive diffusion into the myocytes to prevent complications is still unknown[12].It is imperative to start carnitine supplementation before organ damage for favourable outcomes[13].During periods of fasting, stress, infection and rigorous exercise, prevention of hypoglycemia by providing intravenous dextrose or uncooked cornstarch are recommended.
Carnitine shuttle defects
In children with CPT-1 deficiency MCT supplementation is essential as MCT can enter the mitochondrial without the carnitine shuttle.In the first year 2-3 g/kg/d of MCT and after infancy, 1-1.25 g/kg/d is recommended[9].The clinical and biochemical features of CACT and CPT-2 deficiencies will mimic each other that can be confirmed by enzyme assay on cultured fibroblasts or genetic mutation analysis.Diet rich in complex carbohydrates, low in fat with MCT and carnitine supplementation during well periods and intravenous glucose, night-time drip-feeding during crisis time will avoid the accumulation of toxic intermediaries.Carnitine shuttle defects and transport defects are the only conditions in which carnitine supplementation is unambiguous.Emerging therapy in CPT-2 deficiency is bezafibrate which is a peroxisome proliferator-activated receptor (PPAR) alpha agonist.The activated PPAR alpha binds to PPAR responsive areas on the DNA and stimulates gene transcription.Thus, bezafibrate improves fatty oxidation in cells with mild deficiency of CPT-2 and not severe phenotypes with no residual activity[14].
VLCAD deficiency
In the early onset severe VLCAD deficiency characterized by predominant cardiac involvement aggressive treatment with intravenous glucose, MCT-based formula and intensive care need to be provided.The intermediate form of VLCAD deficiency with predominant hepatic manifestations ought to avoid prolonged fasting, continue on MCT supplements (20% of total energy) and restrict long-chain triglycerides (LCT) to 25%-30% of total energy.The myopathic form presents mostly in adolescents with episodic symptoms and they require pre-exercise MCT supplements (0.25 to 0.5g/kg).Administering oral glucose just before exercise may be detrimental as it will block gluconeogenesis.Restriction of exercise is not advocated.Carnitine supplementation in VLCAD deficiency is controversial as it may increase the plasma flux of toxic acyl carnitine intermediates causing cardiac arrhythmias and rhabdomyolysis[15].This has been proven by studies on cardiomyocytes derived from skin fibroblasts of VLCAD deficient patients.They showed improvement in electrophysiological abnormalities after exposure to etomoxir (inhibitor of CPT-1) which reduced the production of longchain acyl carnitines[16].Thus, empirically starting carnitine in a child where FAOD is suspected without ascertaining the individual defect can be harmful.Bezafibrate has also been shown to improve the residual enzyme activity in VLCAD deficient fibroblasts however, in severe forms of VLCAD deficiency with no enzyme activity, bezafibrate is not useful[17].Attempts to improve exercise tolerance by administering ester form of hydroxybutyrate as the ketone body donor which can be subsequently oxidized has yielded encouraging results in small studies[18].The outcome of nutritional ketosis on other manifestations and long-term effects is still undetermined.
When VLCAD deficiency is suggested by newborn screening, there are several issues that need to be addressed.First is the accuracy of the test, tandem mass spectrometry is used throughout the world and due to the extreme sensitivity, a large number of heterozygous carriers are detected.Therefore, a positive screen has to be confirmed by enzyme assay in cultured fibroblasts or mutation analysis.The next dilemma is regarding the management of asymptomatic VLCAD deficiency as many of them continue to remain asymptomatic and the need for aggressive therapy is contentious.On the other hand, some may develop myopathic form later[19].Hence, in asymptomatic newborns continuation of breastmilk is advised if CPK and transaminases are normal.Whereas high MCT low LCT formula feeds are recommended if CPK or transaminase elevation is present[20].
MCAD deficiency
MCAD deficiency is the most common FAOD and manifests with hepatic and neurological features.Management revolves around avoidance of fasting, taking complex carbohydrates and during periods of crisis intravenous glucose[21].There is no proven role for carnitine and MCT supplementation.Ammonia scavengers are added in cases with severe hyperammonemia and encephalopathy.Since there are no specific therapies compliance to dietary therapy assumes utmost importance.If detected on newborn screening, breastfeeding can be continued with avoidance of prolongation of feeding intervals.In all patients with FAOD, maximum periods of fasting during periods of wellness are as follows: Neonates-3 h, < 6 mo-4 h, 6 to 12 mo-4 h in the day time and 6 h at night, > 1 year-4 h in the day time and 10 h at night[22].
SCAD deficiency
The clinical significance of SCAD deficiency is questionable because of multiple reasons.Children suspected to have SCAD deficiency had hypotonia, epilepsy and developmental delay however, almost all of them remained asymptomatic at followup and some of them were later found to have a different etiology.Newborn screen positive SCAD deficient patients (with elevated C4-carnitine in blood and ethylmalonic acid in the urine) did not develop any abnormality on long-term followup[23].
LCHAD deficiency and TFP deficiency
Irrespective of symptoms, if LCHAD or TFP deficiency is detected in the newborn period, mother's milk has to be replaced with a special formula high in MCT and low in LCT.When complementary feeds are introduced fats should account for 25% to 30% of total energy, MCT should contribute to 20% to 25% of energy from fat and essential fatty acid should be about 3% to 4% and LCT should be restricted to the rest.Essential fatty acids ought to be supplemented with walnut, soy or wheatgerm oil at 3.5 g/d (< 4 mo), 5 g/d (4 to 12 mo), 6 g/d (1 to 4 years) and 10 g/d (> 4 years)[22].Nasogastric feeding can be initiated during mild illness but intravenous glucose is necessary during more severe illnesses.Carnitine supplementation is controversial similar to that in VLCAD defect due to the risk of arrhythmia precipitation.
Docosahexaenoic acid supplementation at 60 mg/d in those < 20 kg and 120 mg/d in > 20 kg have been found to be beneficial in controlling neurological symptoms.
Triheptanoin is a novel therapeutic agent that acts as an anaplerotic metabolite by getting converted to citric acid cycle intermediates for energy production.Triheptanoin is a triglyceride with 7 carbon fatty acids that get metabolized to acetyl CoA and propionyl CoA.When taken at 3 to 4 mL/kg/d it has been shown to improve cardiac functions and reduce metabolic crisis[24].However, making a universal recommendation based on small case series is precarious.The usefulness of triheptanoin has been demonstrated only in LCHAD and VLCAD deficiencies.
Thus, diagnosing and managing FAODs requires a highly nuanced approach and utmost compliance with dietary advice.Supplemental therapy with other agents has to be done after carefully considering the type of defect and the expected difference in the outcome.
UCD
Epidemiology
The incidence of UCD is difficult to deduce as newborn screening programs routinely screen for only arginosuccinate synthetase (ASS) deficiency and arginosuccinate lyase deficiency.The incidence of these two disorders is 1:117000, however, they account for only 30% of all UCDs.Based on newborn screening and natural history studies the combined incidence of all UCDs is estimated to be about 1 in 35000.Ornithine transcarbamoylase (OTC) deficiency accounts for 57%, carbamoyl phosphate synthetase 1 (CPS-1) deficiency for 8% and the others for 1%-2% of all UCDs[25].Though the median age at diagnosis of UCD is 362 d, 25% were diagnosed only in adulthood.Those with late-onset symptoms had more delay in diagnosis of the underlying disorder.However, the patients with severe forms of UCDs may have died before receiving a positive diagnosis.Of 343 UCD cases, 70% were diagnosed after the onset of symptoms, most of whom presented with an acute metabolic crisis[26].Neonatal onset forms of UCDs have a survival ranging from 75% to 90% whereas lateonset forms have survival of > 90%[27].
Urea cycle in brief
The urea cycle metabolizes ammonia into urea (Figure 3).Ammonia is generated in the body from the breakdown of proteins and also from the gastrointestinal tract by bacterial metabolism.The first and the rate-limiting enzyme of the urea cycle is carbamoyl phosphate synthetase 1 which converts ammonia and bicarbonate into carbamoyl phosphate with the aid of NAG.N-acetyl glutamate synthase (NAGS) produces NAG from acetyl CoA and glutamate.NAG activates CPS-1.The second step of the urea cycle involves the synthesis of citrulline from carbamoyl phosphate and ornithine.The first two steps occur in the mitochondria.Citrulline is transported to the cytoplasm by ornithine transporter 1 (ORNT1).The next step is the production of argininosuccinate from citrulline and aspartate by argininosuccinate synthetase.Aspartate is transported from mitochondria to the cytoplasm by citrin.Arginosuccinate lyase breaks down argininosuccinate into arginine and fumarate.The final step is the hydrolysis of arginine to yield urea and ornithine by arginase 1.Ornithine reenters the urea cycle by getting transported into the mitochondria by ORNT1 and urea gets excreted in the urine.Hyperammonemia is universal in all UCD and the manifestations are predominantly neurological.Liver involvement has been described in almost all the UCDs and they range from incidentally detected hepatomegaly to fatty liver, neonatal cholestasis, acute liver failure and cirrhosis[28].
Hurdles in diagnosing UCDs
UCDs can present in the neonatal period, childhood and rarely in adulthood for the first time.The disease spectrum varies from severely symptomatic form associated with high mortality to asymptomatic form depending on the carrier state and residual enzyme activity.Hyperammonemia has to be interpreted based on the age of the child as neonates normally have ammonia levels of 110 μmol/L whereas that in adults is 35 μmol/L[29].When a child presents with encephalopathy and hyperammonemia, the possible causes are acute liver failure of any cause, urea cycle defect, fatty acid oxidation defect, organic acidemia, congenital lactic acidosis, lysinuric protein intolerance (LPI), carbonic anhydrase VA deficiency and portosystemic shunts.Differentiating each of these causes in an acute setting is challenging.UCDs typically do not have acidosis or hypoglycemia, respiratory alkalosis is more common.FAOD, congenital lactic acidosis and organic acidemia will be accompanied by high anion gap acidosis and hypoglycemia.UCDs can present with acute liver failure in 11% to 29% of cases.Cirrhosis also can occur in a proportion of UCDs especially in late-onset forms making the diagnosis extremely difficult for the unsuspecting clinician.Among the various UCDs, the specific defect can be diagnosed based on the profile of the different metabolites as shown in Figure 4.When citrulline is low with low orotic acid, the possibility is CPS-1 deficiency and NAGS deficiency, but if orotic acid is high OTC deficiency is to be considered.With high citrulline, ASS deficiency is the possibility with low argininoscuccinate and arginosuccinate lyase deficiency with high argininosuccinate.Arginase deficiency would have elevated arginine levels.In ornithine translocase deficiency ornithine and homocitrulline levels will be high.In cases with LPI, episodes are triggered by protein-rich meal akin to UCD but in addition, there will be diarrhea, nausea, abdominal pain, hepatosplenomegaly, cytopenia and increased urinary excretion of lysine.In LPI there is defective transport of lysine, ornithine and arginine, hence, there is secondary dysfunction of UCD due to deficiency of arginine and ornithine.Renal, lung, cardiovascular and immunological systems are also affected in LPI[30].In carbonic anhydrase deficiency, there is impaired production of bicarbonate which is a substrate for CPS-1, propionyl CoA carboxylase, pyruvate carboxylase and 3-methyl crotonyl CoA carboxylase.Hence, apart from hyperammonemia, there will be lactic acidosis, ketonuria, hypoglycemia, respiratory alkalosis and metabolic acidosis[31].Portosystemic shunts can be diagnosed with ultrasound Doppler or computed tomography.These children would also have hypoglycemia with hyperinsulinemia, high blood galactose levels, hepatopulmonary syndrome[32].
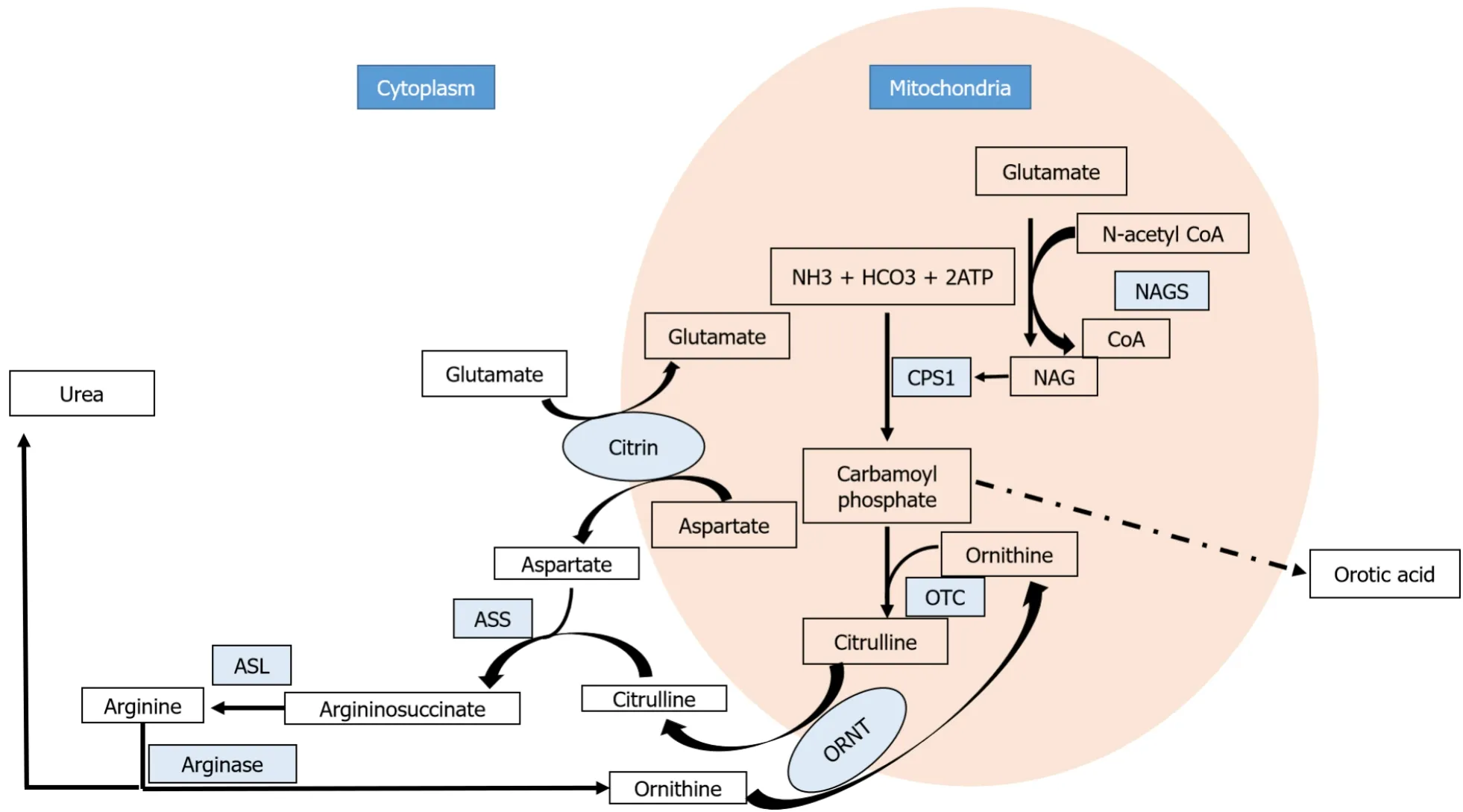
Figure 3 Urea cycle.
Thus, in a child with hyperammonemia, the initial set of investigations should include liver function test, prothrombin time, abdominal ultrasound with Doppler, arterial blood gas, lactate, blood glucose, urine ketones, plasma amino acids, urine organic acids and acylcarnitine profile.Probable diagnosis can be reached with these investigations which can be further confirmed by enzyme analysis or genetic mutation analysis.
Management dile mmas
The cornerstone of the management of UCDs is ammonia clearance and reverse catabolism.In the acute phase, intravenous sodium benzoate (250 mg/kg bolus followed by 250-500 mg/kg/d or 5.5 g/m2/d in those above 20 kg) or sodium phenyl acetate are recommended.For the rapid reduction of ammonia, hemodialysis is advisable especially if the ammonia level is higher than 500 μmol/L or if there is no reduction after 3 to 6 h of starting ammonia scavengers[33].Preparedness for dialysis support is mandatory while waiting for a response from ammonia scavengers.Some children may develop relapse after discontinuation of dialysis hence, continuous hemodiafiltration is preferred.In a severely symptomatic neonate with very high ammonia values hemodiafiltration is the only effective option as peritoneal dialysis is less effective, but performing hemodialysis in a neonate is a formidable task and has to be performed in centers with expertise[34,35].Arginine supplementation (250-400 mg/kg bolus followed by 250 mg/kg/d) has to be given in all UCDs except arginase deficiency.Citrulline supplementation can also increase ammonia excretion through the urea cycle.Oral administration of N-carbamyl glutamate activates CPS-1 similar to NAG.Catabolism reversal is best achieved by the stoppage of protein intake transiently while supplementing carbohydrates and lipids (after ruling out FAOD).However, prolonged discontinuation of proteins would stimulate endogenous protein break-down and hence, 0.1 to 0.3 g/kg/d of proteins have to be restarted from day 2[36].Though the approach seems quite simple there are several practical issues that need to be addressed during the acute presentation.
Acute presentation
Ammonia gets metabolized to glutamine in astrocytes in states of normalcy.When ammonia levels are very high the intracellular glutamine concentration in astrocytes increases.The osmotic effect of glutamine contributes to astrocyte swelling and cerebral edema.Fluid administration has to be diligent especially because ammonia scavengers are given in a large volume of fluid.In addition, massive fluid shifts can occur with sodium load when sodium benzoate and sodium phenyl acetate are administered.Hypokalemia and hyperchloremic metabolic acidosis are also known side effects[37].Thus, close monitoring of electrolytes is recommended.Intravenous glucose infusion aimed at reversing catabolism can result in hyperglycemia which can further worsen cerebral edema.The use of insulin has to be considered in selected situations with close blood sugar monitoring.While the restriction of protein intake in the first 24 h is strongly agreed upon, the use of essential amino acid (EAA) infusion on the first day of decompensation as a nitrogen source to promote anabolism has been used in many centers.Acute hyperammonemia promotes branched-chain amino acid (BCAA) catabolism and ammonia scavengers will also deplete the BCAA pool, hence BCAA supplementation is beneficial[38,39].Enteral feeding has to be always preferred over the parenteral route whenever possible.Arginine and citrulline promote ureagenesis however, administering arginine without establishing the type of UCD can be dangerous as it is contraindicated in arginase deficiency.Arginine is the precursor of nitric oxide and hence, has to be given with caution if the child has vasodilatation and hypotension.Since ammonia is extremely toxic to the brain irreversible neurological damage can occur.The criteria to be used to change treatment strategy from curative to palliative is still debatable.Presence of hyperammonemia for more than 3 d or if the absolute value of ammonia is more than 1000 μmol/L are regarded as poor prognostic factors for favourable neurological outcomes[33].Electroencephalogram and magnetic resonance imaging will assist in predicting the potential of recovery.Continuous electroencephalogram monitoring in neonates with hyperammonemia and deep coma can detect clinically unapparent seizures[40].The decision to continue or withdraw therapy in such situations must be taken after discussion with the family.
Asymptomatic patients
In an asymptomatic neonate with an affected sibling who had a neonatal presentation, glucose infusion ought to be started with 6 hly ammonia measurements.Protein-free feeds and low-dose sodium benzoate (50 mg/kg/d) can be initiated if the baby is asymptomatic after 4 h.If ammonia is consistently < 80 μmol/L, breastfeeds can be introduced after 24 h.If the sibling had a late-onset phenotype, breastfeeding can be given with periodic monitoring of ammonia and symptoms.The relevance of newborn screening in detecting UCDs is still ambiguous.On one hand, the severely affected babies become symptomatic even before the screening results would be available and on the other many positive cases may be detected who may remain asymptomatic throughout.The instability of glutamine and poor accuracy of citrulline levels make them poor markers for screening[41].Arginine and Arginine succinoacetate levels are used in some countries for newborn screening however, many false-positive cases would add to the burden of further workup, parental stress and close monitoring[42].The next dilemma is about managing asymptomatic carriers especially in those with OTC deficiency.Females with severe form may become symptomatic in 15% to 20% of cases.Whether it is prudent to restrict proteins in the initial few years of life is a matter of debate.Relying entirely on genetic studies would be precarious as mosaicism due to lyonization can affect OTC activity.Measuring orotic acid levels after allopurinol administration can detect female carriers with better certainty and it is less dangerous than protein loading tests[43].
Chronic therapy
Long-term management goals revolve around preventing hyperammonemia, ensuring normal growth and development and dealing with periods of anticipated decompensation.Balancing adequate calorie intake with restriction of proteins requires very meticulous planning as the energy required for normal growth in UCD patients is the same as for normal children.The other practical difficulty in achieving optimal protein intake is the food refusal, protein aversions, nausea, vomiting and early satiety in children with UCD that would result in chronic protein deprivation[44].Defining protein restriction is problematic as the requirement changes with age and state of catabolism.Recommended protein intake in normal children varies from 1.8 g/kg/d during early infancy to 0.8 g/kg/d in later childhood which would again increase during periods of crisis, pregnancy and lactation[45].When protein intake in children with various UCDs with good metabolic control was assessed it was found that most of them consumed 1 to 1.8 g/kg/d of protein[38].However, some centers prescribe 0.7 g/kg/d proteins[46].Severe restriction of proteins will affect growth and cause deficiency of vitamins and minerals as well.EAA deficiency is a real problem due to multiple factors like highly restrictive diet, food aversions and EAA depletion by ammonia scavengers.Of the total protein intake, 20% to 30% can be provided as EAA which can be increased to 50% in disorders such as arginase deficiency.Another unanswered question is whether to administer EAA in addition to natural protein or replace a part of natural protein.BCAA supplementation also has wide variations across centers and the optimal dose is yet to be determined.Maintaining enough protein intake to ensure normal growth, prevent endogenous catabolism and also avoid hyperammonemia is akin to a tight rope walk.An absolute value cannot be recommended as requirements vary with age and clinical situations.Natural proteins, as well as EAA supplements, should be divided and consumed in a day.The last meal should provide 25% of total energy to avoid overnight catabolism.
Sodium benzoate or phenylacetate ought to be given with meals and adequate fluids to reduce mucositis and gastritis.BCAA depletion and hypokalemia can occur with chronic therapy also.Other side effects include loss of taste, unpleasant body odour and menstrual disturbance.When using arginine, plasma levels need to be maintained at 70 to 120 μmol/L.Citrulline is the precursor of arginine and both need not be given together.However, the advantage of one over the other in specific disorders is yet to be understood.Monotherapy with carbamyl glutamate is sufficient for NAGS deficiency hence, it is important to differentiate between CPS-1 and NAGS deficiencies which will mimic each other clinically and biochemically.Therefore, enzyme analysis or genetic testing is mandatory for delineation.Secondary carnitine deficiency can be there in children with UCDs and only in severe deficiency do carnitine supplements need to be given.The role of gut metronidazole or neomycin to reduce ammonia load from the colon is not convincing and is not routinely recommended[33].
Monitoring
Given the problems in the measurement of ammonia levels glutamine has been used in monitoring metabolic control with a target of < 1000 μmol/L.Glutamine has been considered as a portent of hyperammonemia.However, there are many inherent issues with using glutamine as the ideal metabolite for monitoring.Preventing permanent neurological damage is the ultimate objective of treating UCDs.Plasma glutamine levels do not correlate with brain glutamine concentration and may not indicate the actual exposure of the brain to ammonia.The ideal cut-off of glutamine as a measure of metabolic control is also uncertain as many children with glutamine well above 1000 μmol/L have not had decompensation.Measuring the levels of ammonia scavengers in blood has also been tried to avoid toxicity but the varying peaks and nadirs make this an unreliable method[47].Apart from biochemical monitoring, developmental and behaviour assessment form a major part of routine evaluation.Periodic magnetic resonance imaging is necessary to predict objective outcomes.
Liver transplantation
Liver transplantation has the potential to be curative in UCDs and is the key to attaining freedom from strict dietary plans.Quality of life and survival improves significantly and since the synthetic functions of the liver are preserved in most cases, long-term outcomes are good.Enzymes of the urea cycle are also expressed in the intestine, kidneys and brain which have physiological functions.Hence, it may be improper to call liver transplantation completely curative.Despite liver transplantation, plasma citrulline levels may be deficient in those with OTC and CPS-1 deficiencies who will require continuing supplementation with citrulline.To achieve maximum benefits from liver transplantation it is crucial to perform it before any irreversible neurological damage occurs.Survival with intact neurological functioning is better with late-onset forms.Thus, in early-onset forms, ideal age for liver transplantation is before 1 year.With the benefits come the challenges of performing transplantation in an infant which demands surgical and critical care expertise[48].When UCDs present with acute liver failure or rarely with end-stage liver disease liver transplantation becomes the only therapeutic option.The timing of referral for transplantation in those with liver dysfunction (not yet causing liver failure or decompensation) is ambiguous as the UCD patients are also eligible for MELD exception points.The effect of medical management on the overall liver functioning in this situation is unknown.Hepatocyte transplantation and auxiliary partial orthotopic liver transplantation are emerging therapies in UCDs[49].
Evolving therapies for UCDs made a foray into clinical practice with much fanfare as these are ideal disorders for gene therapy[50].However, fatal inflammatory complications in a subject related to the immunogenicity of the viral vector paused any further experimental therapies for a long period of time[51].Enzyme replacement therapy in arginase deficiency is not yet a realistic goal in humans[52].
CONCLUSION
Despite making an accurate diagnosis of the disorder management of FAOD and UCD is dotted with ambiguity with respect to the available pharmacological options, strict dietary control and monitoring.We have come a long way in our understanding of metabolic liver diseases but fine-tuning management for optimum results is an ongoing exercise that requires expertise and further research.
杂志排行

World Journal of Hepatology的其它文章
- Hepatitis C virus: A critical approach to who really needs treatment
- Current aspects of renal dysfunction after liver transplantation
- Hepatitis C: Problems to extinction and residual hepatic and extrahepatic lesions after sustained virological response
- Metabolic and nutritional triggers associated with increased risk of liver complications in SARS-CoV-2
- Recent updates on progressive familial intrahepatic cholestasis types 1, 2 and 3: Outcome and therapeutic strategies
- Targets of immunotherapy for hepatocellular carcinoma: An update