Recent updates on progressive familial intrahepatic cholestasis types 1, 2 and 3: Outcome and therapeutic strategies
2022-02-12SeemaAlamBikrantBihariLal
Seema Alam, Bikrant Bihari Lal
Seema Alam, Bikrant Bihari Lal, Department of Pediatric Hepatology, Institute of Liver and Biliary Sciences, New Delhi 110070, India
Abstract Recent evidence points towards the role of genotype to understand the phenotype, predict the natural course and long term outcome of patients with progressive familial intrahepatic cholestasis (PFIC).Expanded role of the heterozygous transporter defects presenting late needs to be suspected and identified.Treatment of pruritus, nutritional rehabilitation, prevention of fibrosis progression and liver transplantation (LT) in those with end stage liver disease form the crux of the treatment.LT in PFIC has its own unique issues like high rates of intractable diarrhoea, growth failure; steatohepatitis and graft failure in PFIC1 and antibody-mediated bile salt export pump deficiency in PFIC2.Drugs inhibiting apical sodium-dependent bile transporter and adenovirus-associated vector mediated gene therapy hold promise for future.
Key Words: Genotype; Biliary diversion; Gene therapy; Liver transplantation
INTRODUCTION
Progressive familial intrahepatic cholestasis (PFIC) is estimated to affect 1 in 50000-100000 births[1].With improved awareness about the various presentations of these bile transport disorders, we now know that the clinical presentation can vary from early-onset severe liver disease to episodic late-onset occurrence triggered by an external stimulus.Figure 1 describes the various transporters which are involved in different types of PFIC.This article will focus on the PFIC types 1, 2 and 3.Table 1 depicts the phenotype and genotype of these three types of PFIC.
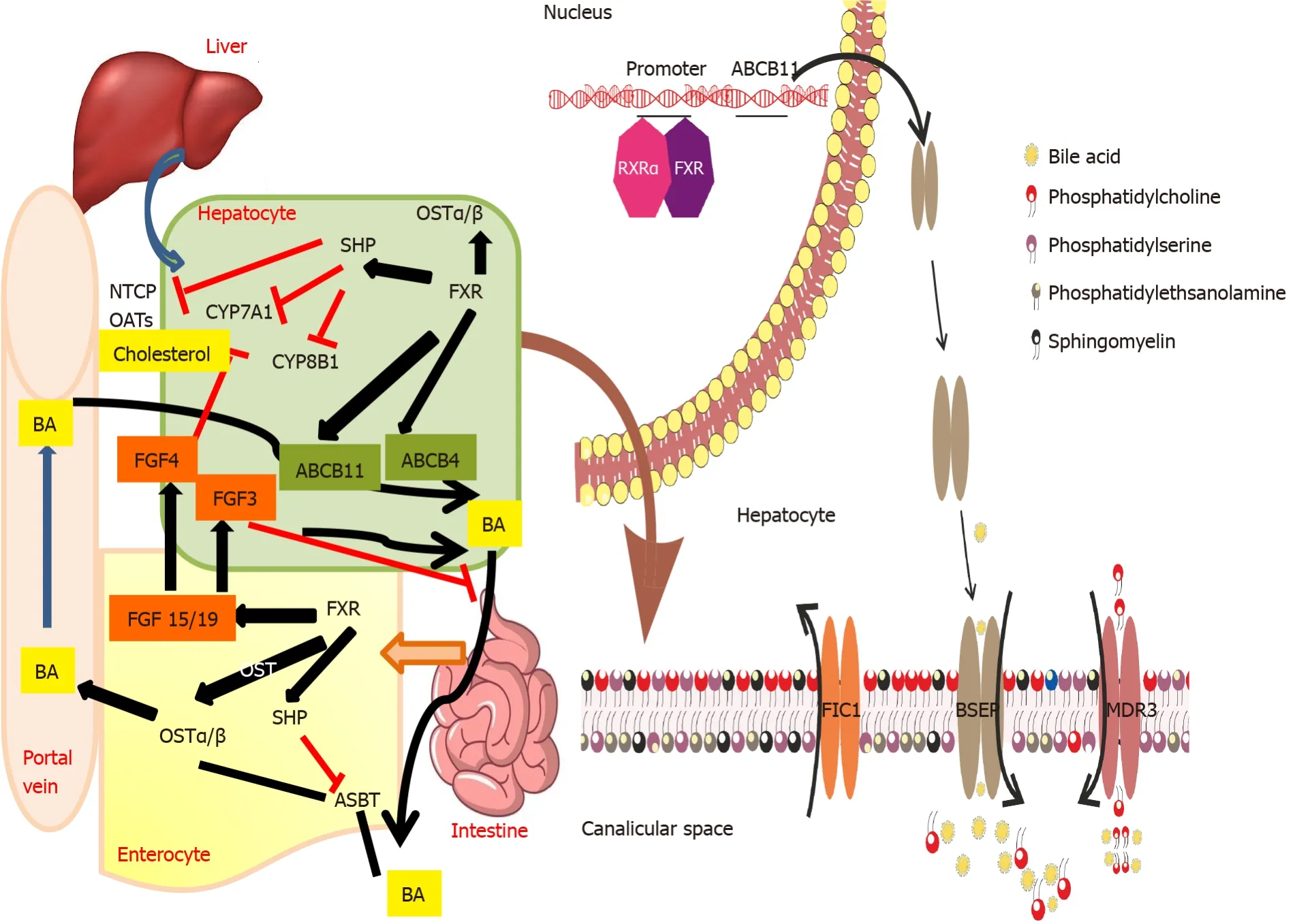
Figure 1 Bile acid secretion and transport from the hepatocyte across the canalicular membrane and enterohepatic circulation: Role of various transporters involved in transport of bile salts and phospholipids across the canalicular membrane.Enterohepatic circulation of bile salts and their regulation via farnesoid X receptor - fibroblast growth factor 15/19 is also depicted.

Table 1 Genotype, phenotype and histopathological differentiation of the various types of progressive familial intrahepatic cholestasis
FAMILIAL INTRAHEPATIC CHOLESTASIS 1 DEFICIENCY
Prevalence of familial intrahepatic cholestasis 1 (FIC1) deficiency is 10.4%-37.5% amongst the cholestatic disorders[2].Earlier known as Byler’s disease, seen predominantly in children of Amish descent, this disease is now known to occur in all racial and ethnic groups.
Pathogenesis
FIC1 is encoded by the ATPase phospholipid transporting-8B1 gene (ATP8B1), located on chromosome 18 and responsible for maintaining the asymmetric distribution of phospholipids across the lipid bilayer by the flippase movement of phospholipids from the outer lipid leaflet of the canalicular membrane to the inner lipid leaflet.Earlier it was believed that FIC1 was responsible for the flippase movement of phosphatidyl-serine, but it is now proven that the preferred substrate is phosphatidylcholine[3].Phosphatidyl-choline helps in protecting the canalicular membrane from hydrophobic bile acids (BA).Hence, in the absence of FIC1, proteins in the canalicular membrane such as the bile salt export pump (BSEP) can have impaired function.ATPbinding cassette subfamily B member 4 (ABCB4) and ATP8B1 maintain lipid asymmetry which is important for the integrity of the canalicular membrane.When ABCB4 flops the phosphatidyl-choline, there is destabilization of the canalicular membrane which is counteracted by the flippase activity of FIC1.Hence by maintaining the integrity of the canalicular membrane, FIC1 maintains the function of multidrug-resistant type 3 (MDR3) which is also known as ABCB4[4].The mechanism by which loss in ATP8B1 leads to cholestasis is poorly understood, but it has been proposed that an abnormal lipid packing (due to the loss in ATP8B1 flippase activity) makes the outer leaflet of the canalicular membrane susceptible to bile salt-mediated extraction of cholesterol and phospholipids.This results in reduced activity of ABCB11, a major bile salt exporter that has also been linked to PFIC2[5-7].Further, the lower expression of FIC1 down-regulates farnesoid X receptor (FXR) expression, encoded by the NR1H4 gene.FXR has a dual action.FXR in hepatocytes regulates the secretion of bile through BSEP and FXR in enterocytes regulates bile salt reabsorption through apical sodium-dependent bile acid transporter (ASBT)[8].
FIC1 is expressed in a variety of tissues, including the canalicular membrane of hepatocytes, apical membrane of cholangiocytes, brush border of enterocytes and cochlear hair cells[9].Hearing loss is attributed to defects in the composition of membranes of inner ear cilia.Knockdown ATP8B1 in Caco-2 cell model of intestinal epithelium leads to an unorganized apical actin cytoskeleton and post-transcriptional defect in apical protein expression.This prevents the movement of the ATP8B1 into the apical membrane further causing inhibition of ASBT through the intestinal FXR, eventually resulting in bile malabsorption and diarrhea[8].FIC1 is also expressed in the pneumocytes where ATP8B1 inhibits cardiolipin.Hence deficiency of ATP8B1 increases the cardiolipin which can disrupt surfactant function within pulmonary alveoli and increase the risk for pneumonia[10].ATP8B1 deficiency is also associated with down-regulation of cystic fibrosis transmembrane conductance regulator (CFTR) and increased sweat chloride test in 15% of patients.
Clinical and laboratory presentation
Based on the patients’ clinical, laboratory profile, and liver histology, the presentation of ATP8B1 deficiency ranges from a severe (earlier known as PFIC1) to a milder phenotype [earlier known as benign recurrent intrahepatic cholestasis (BRIC)][11].
Severe ATP8B1 deficiency presents in children with refractory cholestasis (typically beginning in early infancy) manifesting as jaundice, severe failure to thrive (disproportionate to the degree of cholestasis), delayed puberty and fat-soluble vitamin deficiency (e.g.,coagulopathy of vitamin K deficiency).The jaundice can often beintermittent.The liver disease is progressive causing secondary biliary cirrhosis and end-stage liver disease.Pruritus is often disproportionate to hyperbilirubinemia but correlates with the serum BA levels[1].Symptoms begin in the first month of life in 15% of patients and by 3 mo of age in 61% of patients with PFIC1[12].Undernutrition is generally responsible for poor growth.However, proportionate growth failure and delayed puberty together suggests a likely systemic manifestation of ATP8B1 deficiency.BA is high and serum cholesterol is characteristically low.ATP8B1 deficiency is associated with extrahepatic manifestations such as hearing loss, pancreatitis (8%) or pancreatic exocrine insufficiency, kidney stones, resistance to parathyroid hormone and diarrhea (61%) not attributable to fat malabsorption[9,13].The severe form of ATP8B1 deficiency may be associated with congenital hypothyroidism (both clinical + subclinical) which can be treated[14].Coarsely granular canalicular bile may be found on electron microscopy which may improve after the administration of ursodeoxycholic acid (UDCA).A recent multi-centric study included 130 patients with pathological ATP8B1 mutations and low-gamma-glutamyl transpeptidase (GGT) cholestasis phenotype[15].Among these patients, 15 initially presented as BRIC that later evolved into a severe FIC1 phenotype.Age at first presentation to the tertiary center was 0.6 (0.3-2.2) years, illustrating an early onset of disease.Hepatocellular carcinoma (HCC) was not seen in any of the patients, either before transplantation or in the explants.
Mild ATP8B1 deficiency: Episodic cholestasis beginning later in childhood, adolescence or young adulthood usually rules out severe disease.The presentation may include pruritus without jaundice or presence of mild jaundice.Episodes may last from weeks to months and symptom-free intervals may vary from months to years.The intermittent disease pattern is usually associated with different or unknown triggers which may include fever or any other inter-current illness, drugs (such as contraceptives), malignancy, hormonal changes or pregnancy.BA is high and serum cholesterol is low during symptomatic periods.Histopathological findings during the symptomatic periods may be milder, yet similar to that seen in severe form.
Genotypic variation
In a recent study with 18 potential disease-causing mutations in ATP8B1, 14 patients (4 homozygous, 6 compound heterozygous, 4 heterozygous) were below 18 years of age and had a median age of 0.75 years[16].Overall, 28 different genetic variants were identified including two common single nucleotide polymorphisms (SNPs) (p.R952Q and c.3531+8G>T)[16].These variants have been described earlier in European patients with intrahepatic cholestasis of pregnancy (ICP)[17] and pancreatitis[13].Pathogenic variants with underlying severe ATP8B1 deficiency are likely to be fully penetrant but some of the milder variants may not become symptomatic even in adulthood.The p.Ile661Thr pathogenic variant, which is frequently detected in people with a mild disease of European descent, appears occasionally to be non-penetrant.However, it is also found in compound heterozygous form in persons with severe disease.A multicentric study on FIC1 patients further classified the patients based on the presence of predicted protein-truncating mutations (PPTMs) (i.e.,splice site, frameshift due to deletion or insertion, nonsense, duplication)[15].FIC1-A patients harbored no PPTMs, FIC1-B patients had one PPTM, and FIC1-C patients manifested two PPTMs.Patients with FIC1-C genotype presented to the referral center at 0.4 (0.2-0.7) years, which is significantly earlier as compared to FIC1-A [0.8 years; interquartile range (IQR): 0.4-3] and FIC1-B (0.9 years; IQR: 0.4-2.7) respectively (P= 0.004).At presentation, levels of BA, aminotransferases, and GGT were comparable among the three genotype groups.FIC1-B patients had the highest bilirubin levels.Half the cases of FIC1-A were receiving UDCA at presentation but did not show a significant improvement biochemistry in comparison to the other patient groups.
Natural history and outcome
The majority of the severe variants of ATP8B1 deficiency will have disease onset within infancy.Some may experience episodes of severe cholestasis followed by disease-free intervals with eventual persistence of cholestasis.Malnutrition and fatsoluble vitamin deficiencies can cause significant morbidity, if left untreated.Features of chronic liver disease and portal hypertension may develop leading to cirrhosis by the end of the first decade of life but variations have been noted within families[15].Without surgical intervention, end-stage hepatic failure and death usually occur in the second decade of life.In some of those with mild disease, clinical monitoring may reveal a shift in the clinical spectrum from mild intermittent disease to more persistent cholestasis and fibrosis on the follow-up biopsies[2].The Natural course and Prognosis of PFIC and Effect of biliary Diversion (NAPPED) consortium data revealed that 8 of 130 patients (6%) died before liver transplantation (LT), of which 3 had undergone surgical biliary diversion (SBD)[15].Survival analysis showed that at 18 years of age, 44% of patients were alive with their native liver.Three-fourths of those alive with their native liver had undergone SBD by 18 years and a significantly lower percentage of patients with a FIC1-C genotype underwent SBD (FIC1-A 65%, FIC1-B 57%, FIC1-C 45%;P= 0.03).Native liver survival (NLS) was comparable between the three genotype sub-groups at 10 years of age (FIC1-A 67%, FIC1-B 41%, FIC1-C 59%;P= 0.15) with or without SBD.However, the proportions of total patients alive with the native liver at the age of 10 years were lower in patients without SBD than in patients who had undergone an SBD during follow-up.
BSEP DEFICIENCY
BSEP is a liver-specific transporter involved in actively transporting monovalent bile salts out of hepatocytes into biliary canaliculi against a concentration gradient.BSEP is encoded by the ABCB11 gene located on chromosome 2q31.Homozygous or compound heterozygous mutations in this gene results in PFIC2[18].
Pathogenesis
Chenodeoxycholic acid (CDCA) and the Retinoid X Receptors (FXR) α-heterodimer together activate the ABCB11[18].Increasing bile acid concentration in the hepatocytes stimulates greater synthesis of BSEP to maintain equilibrium.There are over 200 mutations of ABCB11 causing PFIC2[18].Common mutants that cause retention of the protein in the endoplasmic reticulum are G238V, D482G, G982R, R1153C, R1286Q, and ΔGly.The mutant protein that is retained and translocated out of the endoplasmic reticulum is then broken down by proteasomes[19].Intrahepatic accumulation of bile salts and inflammation promote carcinogenesis through genomic modifications[19].Metabolomic studies have shown that ABCB11 knock-out mice have impaired mitochondrial fatty acid β-oxidation with the resulting increase in reactive oxygen species that might exacerbate the liver injury[19].
Clinical and laboratory presentation
BSEP deficiency is the most common subtype of PFIC (with an estimated incidence of 1 in 50000 to 1 in 100000)[8].It accounts for 37.5%-90.9% of cholestatic patients in the 9 studies that were analyzed in a systematic review[2].Symptoms appear in the first month of life in 44% and by 3 mo of age in 72%[12].Some patients with BSEP deficiency also present with early signs of vitamin D deficiency (rickets 3%-22%), vitamin K deficiency (bleeding 8%) or cholelithiasis (28%)[8,12].High serum alanine aminotransferase and alpha-fetoprotein levels, early liver failure, cholelithiasis, HCC, very low biliary BA concentration, and negative BSEP canalicular staining suggesting PFIC2 (Figures 2A and 2B)[12].In infancy, PFIC2 has mild to severe portal and lobular fibrosis with bridging fibrosis.Beyond infancy, advanced portal and lobular fibrosis are observed[2].Early progression to HCC is described[19].For more details see Table 1.
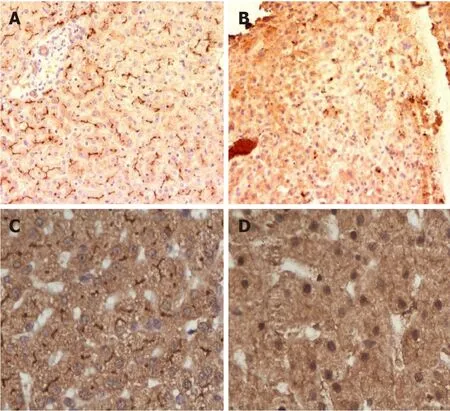
Figure 2 Immunohistochemistry of progressive familial intrahepatic cholestasis type 2 and 3.
Genotypic variation
Of the 252 patients with cholestatic liver disease screened for ABCB11, 88 (34.9%) were identified with at least one disease-causing BSEP mutation with the median age of disease onset being 0.75 years[16].64.3% had no disease-causing mutation but at least one BSEP SNP.Amongst the common SNPs, p.A1028A was found in 197 of 252 families and p.V444A in 204 of 252 families of this cohort.The most common PFIC-2 mutations were p.E297G, p.D482G, and p.N591S.Apart from PFIC2, ABCB11 mutations cause BRIC2, ICP, contraceptive induced cholestasis and drug-induced cholestasis[16].Four potential ABCB11 mutations and a donor splice site mutation (intron 19) were identified in 147 women with ICP[17].Please also see the section on BRIC2 and ICP.
A global consortium on BSEP deficiency categorized the mutations as BSEP1 [p.D482G (c.1445A>G) or p.E297G (c.890A>G) mutation], BSEP2 (at least 1 missense mutation, not p.D482G or p.E297G) or BSEP3 (mutations leading to a predicted nonfunctional protein)[20].Patients with BSEP1 genotype (p.D482G and p.E297G) have residual BSEP functionality and thus have a milder phenotype as compared to patients exhibiting BSEP2/BSEP3 genotype.There is wide geographical variations in genotype with the commonest mutations found in a study from Shanghai being c.145C>T (p.Gln49Ter) and c.2594C>T (p.Ala865Val)[21].Severe phenotypes are often associated with mutations leading to premature protein truncation or failure of protein production.Insertion, deletion, nonsense and splicing mutations exhibit little or no detectable BSEP at the hepatocyte canalicular membrane.
Natural history and outcome
PFIC2 is likely to have more severe lobular injury, portal fibrosis and inflammation than PFIC1.Hepatocellular necrosis and giant cell transformation are also much more pronounced in PFIC2 than in PFIC1 and may persist with time.Majority of homozygous or compound heterozygous mutations present as severe disease in infancy with progressive liver disease which requires LT.Despite treatment, 76%-100% of PFIC2 continue to have pruritus with progression to severe liver disease and this may occur as early as 7 mo of age.Advanced portal and lobular fibrosis are seen in children beyond infancy[12].Among patients undergoing LT, liver failure and/or HCC were detectable in about 60%.Liver samples obtained from 10 children with HCC aged 13-52 mo showed that 9 had evidence of BSEP deficiency and/or mutations in ABCB11.Recently, HCC and/or cholangiocarcinoma was described in 15% of those with PFIC2.Hence, close monitoring of these children particularly those carrying 2 null mutations is essential[22].Children with transient neonatal cholestasis harbour non-null variants, express immune-histochemically detectable BSEP and have better outcomes[21].
The global NAPPED consortium reported that 23.1% PFIC2 had undergone SBD and 46.1% had undergone LT[20].In total, 16 (6%) [BSEP1:n= 3/72 (4%), BSEP2:n= 8/136 (6%), BSEP3:n= 5/56 (9%)] died prior to LT at a mean age of 1.6 years.NLS was 32% at 18 years of age.Five patients underwent LT beyond 18 years of age (19.6-27.5 years).Patients with mutations categorized as BSEP1 had better long-term outcomes than those with BSEP2 or BSEP3, with a median NLS of 20.4 years, 7 years and 3.5 years respectively.The incidence of HCC increased with the genotype severity from 4% in BSEP1 to 7% in BSEP2 and 34% in BSEP3.HCC occurred in 3% of patients who had SBD as against 7% who had not received SBD[20].
MDR3 DEFICIENCY
Pathogenesis
This autosomal recessive condition is caused by mutations of the MDR3 glycoprotein, which is coded by the ABCB4 gene located on chromosome 7q21.The gene consists of 27 coding exons spanning-74 kb[23].ABCB4/MDR3 P-glycoprotein consists of six intracellular domains and six extracellular loops separated by twelve transmembrane domains.The protein contains two intracytoplasmic ATP-binding domains, also known as nucleotide-binding domain.The nucleotide-binding domains provide energy for the trans-membrane transfer of the substrate against the concentration gradient.The trans-membrane domains in turn provide specificity for the substrate.A study on fetal liver demonstrated that ABCB4 mRNA levels are 16-fold lower compared to the normal adult liver with only faint and focal canalicular MDR3 immunostaining suggesting that ABCB4 normally develops late in gestation or possibly in the postnatal period.The protein is present in the canalicular membrane of hepatocytes and transports phosphatidyl-choline from the hepatocytes to the bile canaliculi.Phosphatidyl-choline is a key component of micelles which keeps the cholesterol in soluble form thus protecting the cholangiocytes from damage.In presence of abnormal MDR3, phosphatidyl-choline is not transported across to the bile canaliculi leading to abnormal micelle formation.Inadequate micellar formation results in presence of insoluble bile salts and cholesterol in the biliary canaliculi which damage the cholangiocytes.Cholesterol is more likely to crystallize into stones, damaging liver structures by obstructing small bile ducts.
Clinical and laboratory presentation
These patients present with a wide spectr um of manifestations ranging from transient neonatal cholestasis, episodic cholestasis, gall stones, cirrhosis to end-stage liver disease[24,25].The patients with PFIC3 have elevated GGT and alkaline phosphatase.Bile salts and cholesterol values can be normal while biliary phospholipids are significantly reduced.Age of the onset of PFIC3 can vary widely from the neonatal period to adulthood.PFIC3 often presents later as compared to PFIC1 and PFIC2, as late as adulthood in some cases[25].The early-onset disease can present with jaundice, pruritus, variceal bleeding (portal hypertension), stunted growth, reduced bone density and learning disabilities[25].The complete absence of canalicular staining of MDR3 protein is associated with mutations leading to a truncated protein form (Figures 2C and 2D).Those with missense mutations have faint or normal MDR3 canalicular expression.The abnormal MDR3 canalicular expression combined with low levels of biliary phospholipids is highly suggestive of MDR3 deficiency.Histopathological findings associated with PFIC3 include primarily portal-based inflammatory infiltrate with marked periportal ductular reaction.They usually progress to biliary-type cirrhosis, lobular disarray, and hepato-canalicular cholestasis.
Genotypic variation
Degiorgioet al[26] analyzed ABCB4 gene mutations in 68 PFIC3 patients and found 29 mutations in the coding region and 10 in the transmembrane domains which involved phosphatidyl-choline translocation.Delaunayet al[27] devised a classification system for the various forms of these mutations: Class I (nonsense mutations that result in a defective synthesis), class II (missense variations that prevent protein maturation), class III (missense mutants resulting in defective protein activity), class IV (unstable variations) and class V mutants (unknown pathogenicity).These classifications are useful in determining potential therapies for patients.Approximately 300 diseasecausing ABCB4 variants have been reported, typically in those with homozygous status who have a rapidly progressive cholestatic liver disease with potentially lifethreatening complications.Those with heterozygous status have less severe manifestations.
Natural history and outcome
Patients with residual phosphatidyl-choline secretion and MDR3 expression (especially those with missense mutations), respond to treatment with UDCA in 70% of cases.LT is reserved for patients with PFIC3 associated with end-stage liver disease.The mean age at LT is 9.6 (2-33) years[28].HCC and cholangiocarcinoma have both been described with PFIC3.
BRIC
BRIC is an autosomal recessive inherited disorder characterized by the intermittent occurrence of severe and recurrent cholestasis[29].BRIC typically appears later than PFIC and is not progressive.Two genetic forms of BRIC are known; BRIC1 is characterized by the mutations of the ATP8B1 gene while BRIC2 presents a mutation in the ABCB11 gene.BRIC shows a non-progressive course since the protein function is only partially impaired[29].Most of them have missense mutations located in less conserved regions of the gene.The global NAPPED consortium reported 11 BRIC2 who later presented with persistent cholestasis and progressive liver disease with pathological genetic variants[20].
Association with other cholestatic presentations
ICP is the occurrence of intense pruritus in a pregnant woman without pre-existent liver disorder; beginning usually in the third trimester and resolving completely after delivery.Elevated levels of BA, aminotransferase and alkaline phosphatase are typically present.Although the disease is benign in the mothers, poor fetal outcomes have been reported.Heterozygous mutations in ABCB4, ABCB11, ATP8B1, ABCC2 and tight junction protein 2 have been associated with ICP[30].While GGT is normal in ABCB11-related forms of estrogen-associated cholestasis, increased GGT levels could serve as a discriminating feature of ABCB4 deficiency.
The associations of heterozygous missense and nonsense ABCB4 mutations in cryptogenic liver fibrosis and biliary cirrhosis have been reported[28,31].The histomorphological phenotype associated with ABCB4 mutations includes portal fibrosis with periportal ductular proliferation.Early diagnosis and therapy with UDCA may slow down fibrosis progression in few cases.Some cases of ABCB4 mutations may also present with chronic cholangiopathy[32].
MDR3 deficiency also leads to gallbladder disease or low phospholipid-associated cholelithiasis (GBD-1/LPAC)[32].There is more evidence that either biallelic or monoallelic ABCB4 defects may cause or predispose patients to a wide spectrum of human liver diseases such as LPAC, small duct sclerosing cholangitis and adult biliary fibrosis or cirrhosis[28].Liver cirrhosis accompanied by vanishing bile duct syndrome (ductopenia) in ABCB4 deficient patients have been reported[33].
ABCB11 and ABCB4 deficiency may predispose to the cholestatic liver injury induced by oral contraceptives, psychotropic drugs, selected chemotherapy drugs, statins, and antibiotics[34].The common variant p.Val444Ala in ABCB11 is associated with an increased risk of drug-induced liver injury[35].Most cases improve on withdrawing the drug and adding UDCA.Recent studies have demonstrated, that ABCB4 mutations (c.504C>T and c.485T>Ain exon 6 and c.2793 frameshift mutation in exon 23) are possibly involved in the development of parenteral nutrition-associated liver disease in premature infants[36].
THERAPEUTIC STRATEGIES FOR PFIC
The holistic management of patients with PFIC includes their nutritional rehabilitation, fat-soluble vitamin supplementation, control of pruritus, prevention of fibrosis progression and LT for those with end-stage liver disease.
Nutritional rehabilitation
Chronic malnutrition due to cholestasis and steatorrhoea affects almost all children with PFIC, especially those with PFIC1[37,38].Apart from cholestasis-related malabsorption, complex interactions of the liver with the hypothalamus-pituitaryadrenal axis in chronic cholestasis have been advocated as one of the mechanisms for growth failure in these children[39].Severe cholestasis leads to deficiency of fatsoluble vitamins (FSV) with decreased bone mineral density, often refractory to FSV supplementation[38].Meticulous nutritional assessment should be performed and documented at the first visit.The goal of nutritional rehabilitation should be to give calories around 125% of the recommended daily dietary allowance for the ideal weight targeting around 180-200 cal/kg/d along with high protein about 2-3 g/kg/d[38].FSV should be supplemented orally: Vitamin A 5000-10000 IU/d; Vitamin D (cholecalciferol) 2000-5000 IU/d; Vitamin E 50-400 IU/d and Vitamin K 5 mg/wk to 5 mg/d[40].Weekly or daily supplementation of FSV, especially vitamin D, is more effective than large bolus doses for the treatment of vitamin D deficiency in children with chronic liver disease[41].Refractory deficiency or those with significant bony changes should be treated with calcitriol at a dose of 0.05-0.2 μg/kg[40].The oral water-soluble liquid formulation of FSV containing vitamins A, D, E and K have shown to be more efficacious than conventional preparation in infants and children with cholestatic liver disease.Medium-chain triglycerides should be supplemented as they are absorbed directly into the portal circulation and are not affected by cholestasis[40].
Treatment of pruritus
Pruritus is the most disabling symptom of PFIC disturbing daily activities, schooling and sleep.A step-up approach of medical therapy is advocated in children with pruritus.SBD is a very useful tool in medical non-responders (Figure 3).Table 2 lists the drugs used for the management of pruritus with their mechanism of action, dose and adverse effects.Apart from medical therapy, trimmed nails, full-sleeve clothingstockings during sleep and well moisturized skin are advocated.Various tools like visual analog scale, Whitington scale or 5D-pruritus score have been used for objective measurement of pruritus and to assess response to medications or surgical therapy[42].
Role of medical therapy
UDCA leads to complete resolution of pruritus in about a third of children with PFIC2[38] and response is mainly dependent on the mutation and resultant residual protein expression[24].Long-term therapy with UDCA has been shown to reverse fibrosis in MDR3 heterozygotes with residual protein expression[43].The role of rifampicin in management of pruritus has been established by meta-analyses (pooled odds ratio of 15.2 for complete/partial relief) as compared to placebo/alternative therapy[44].Bile acid sequestrants are less favourable in children due to non-palatability[38].Spacing is needed from food and other drugs leading to loss of compliance and optimal effect[45].Opioid antagonists can ameliorate pruritus, although with increased incidence of adverse events[44].Sertraline, a selective serotonin reuptake inhibitor is also beneficial for the relief of pruritus.Sertraline has high first-pass metabolism in the liver and should be used with caution.The other drugs with equivocal efficacy for control of pruritus in PFIC include ondansetron, phenobarbitone and antihistaminics[40,41].Newer modalities to treat pruritus of cholestasis that have not been extensively analyzed in studies include albumin dialysis using molecular adsorbent recirculating system, plasmapheresis and endoscopic nasobiliarydrainage[46].Plasmapheresis reduces autotaxin activity thereby reducing lysophosphatidic acid and reducing pruritus in ICP.
Surgical interruption of enterohepatic circulation
Commonly used SBD include partial external biliary diversion (PEBD), ileal exclusion (IE) and partial internal biliary diversion (PIBD).Figure 4 shows a schematic representation of the various diversion surgeries, techniques and their common complications.PEBD, although being the most widely used diversion surgery is associated with a host of stoma-related complications[47-49].Recently, external fistulae have been intubated with gastrostomy devices for intermittent drainage[49].IE[50,51] and PIBD[52,53] are therapeutic options in those with failed PEBD or for better cosmetic results, especially in adolescents.Recent studies on SBD with their outcome and complications have been tabulated in Table 3[51-55].Biliary diversion leads to a relatively higher ratio of cholic acid to CDCA in bile, reduced taurine-to-glycine conjugate ratio, increase in secondary bile acid pool, ultimately causing bile to become more hydrophilic[55].Total biliary diversion involves diverting all the bile flow from the common bile duct to the exterior through a jejunal loop.It is usually reserved for pruritus refractory to PEBD or to prevent and treat allograft steatosis in post LT PFIC1[56].Irrespective of the technique used, biliary diversion surgeries lead to a decrease in the concentration of BA, improvement in pruritus score, growth spurt, histological improvement and even reversal of fibrosis in some cases[38,47].There are no head-tohead trials to compare the efficacy and safety of these techniques.Post SBD, the child should be monitored for improvement in pruritus, changes in growth parameters and for decrease in BA and bilirubin in the serum.Recently, the NAPPED consortium andanother meta-analysis have shown improved NLS in those with common missense mutations (D482G or E297G) and in those with more than 75% reduction in BA post SBD[20,57].Around 18%-39% of children undergoing SBD require LT in 5-10 years of follow-up, mostly those with advanced fibrosis at the time of SBD and those with failure of SBD[51-55].Those with advanced fibrosis do not show a reduction in serum BA with biliary diversion surgeries, hence a liver biopsy should precede the surgery[57].The choice of biliary diversion technique depends on the patient’s preference and experience and comfort of the surgeon/centre.Health-related quality of life is comparable between those undergoing PEBD for PFIC and healthy controls.
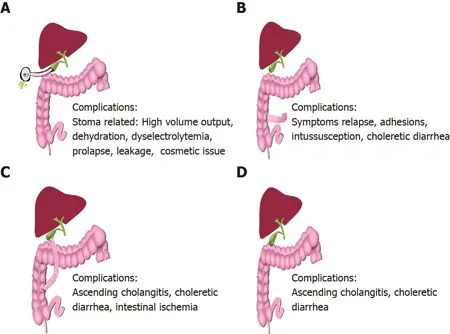
Figure 4 Schematic representation of biliary diversion surgeries.
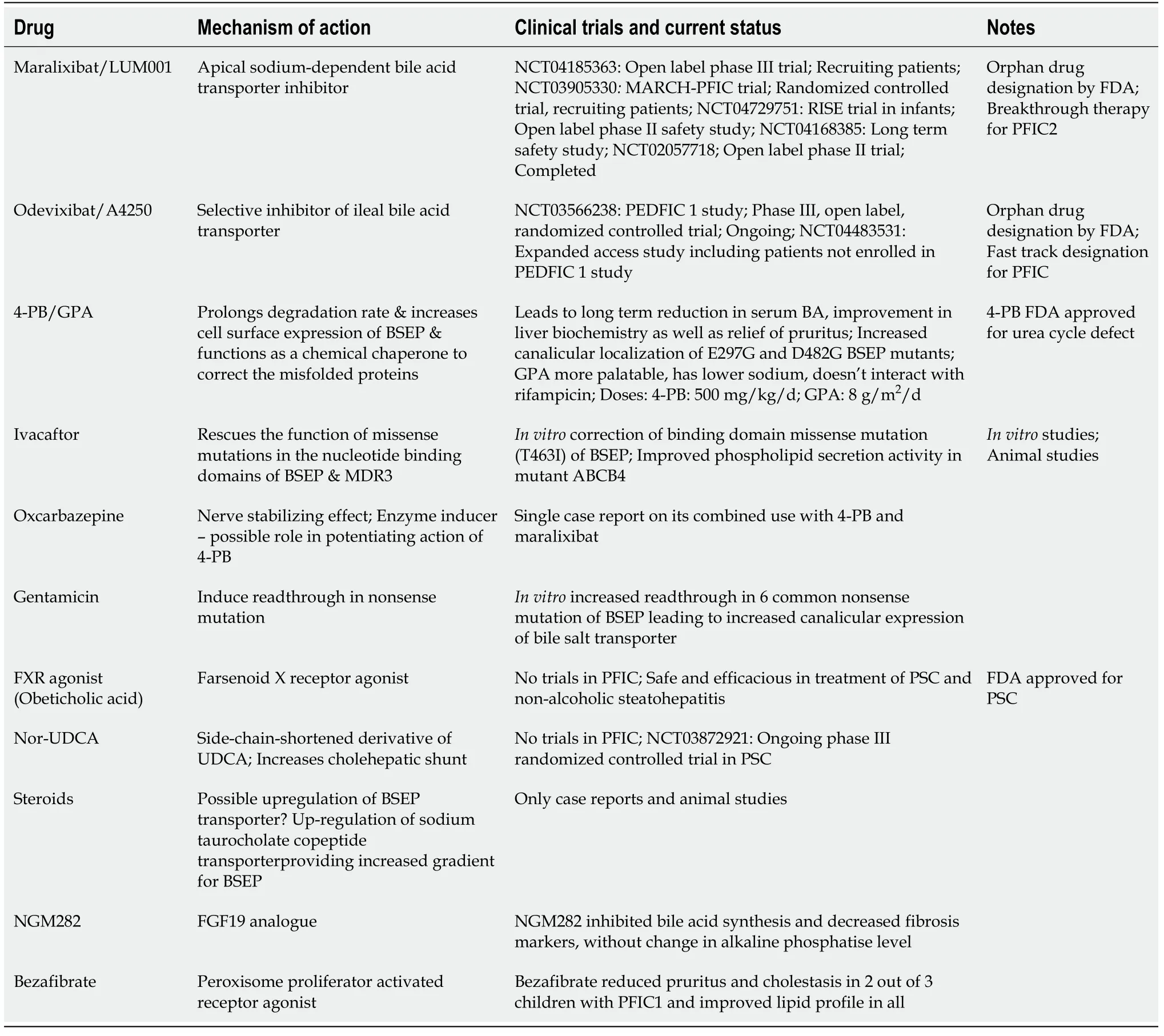
Table 4 Potential novel therapeutic drugs for treatment of progressive familial intrahepatic cholestasis

Figure 3 Algorithm for management of pruritus in children with progressive familial intrahepatic cholestasis.
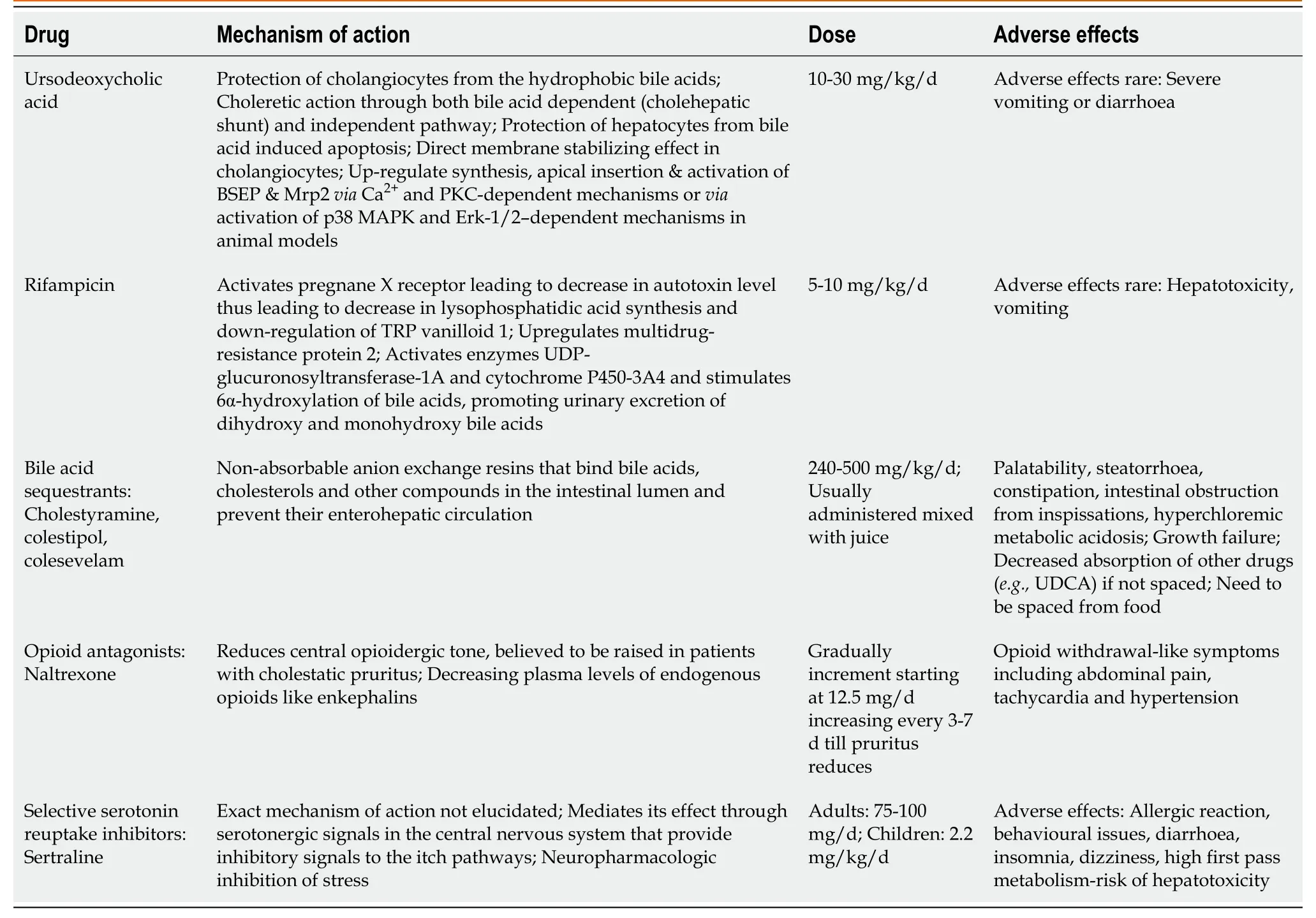
Table 2 Drugs used for control of pruritus in progressive familial intrahepatic cholestasis: Mechanism of action, dose, adverse effects

Table 3 Recent studies describing outcome and complications with biliary diversion surgeries in progressive familial intrahepatic cholestasis
LT
LT is indicated in children with end-stage liver disease, advanced fibrosis and those with refractory pruritus despite biliary diversion surgeries, yet it is fraught with its unique complications and conundrums in children with PFIC[58].PFIC is one of the most common indications accounting for 13.4% of total pediatric LT[59].LT in patients with PFIC2 and PFIC3, where the mutation affects only the liver has resulted in excellent graft and patient survival[37].On the other hand, due to a wide extrahepatic tissue distribution of FIC1 expression, LT for PFIC1 has been associated with a high risk of complications due to the involvement of these extrahepatic sites after LT[60].There are three unique issues related to LT in PFIC: (1) High rate of graft failure, allograft steatosis and diarrhea in PFIC1; (2) Antibody-induced BSEP deficiency leading to graft loss in PFIC2; and (3) Utilization of heterozygous family members as donors in living donor LT program.
Allograft steatosis, refractory diarrhea and graft failure in PFIC1:Post LT in PFIC1, the graft liver starts to secrete and excrete normal amounts of bile salts which can’t be handled by the intestines as the intestinal FIC1 is still defective leading to intractable diarrhea[60].This plausible mechanism for intractable diarrhea is further strengthened by the clinical response to bile adsorption resins and total biliary diversion.FIC1 in theintestine which has a strong correlation to SLC10A2, the ileal sodium/bile acid cotransporter is defective in PFIC1 leading to the impaired enterohepatic circulation of bile salts[61].When the intestine of these patients which are defective in FIC1/SLC10A2 starts receiving a normal amount of BA secreted by the graft liver, it leads to bile acid diarrhea, steatosis and graft fibrosis necessitating re-transplant[60].Horiet al[37] reported chronic diarrhea in 91%, grade 3 steatosis in 72.7% and significant graft fibrosis in 82%; 27% died at a median of 12 years post LT and 3 required re-transplant.Davit-Spraulet al[25] reported extrahepatic complications post LT in 100% of PFIC1 and one-third required re-transplant for massive steatosis.There is a recurrence of steatosis and graft failure even in the second graft[12,60].The genetic analysis predicts the patients likely to develop these complications and thus pre-LT genetic analysis should be considered mandatory for patients with PFIC1[60].Total internal biliary diversion during the time of LT has been described as a modality by the Kasahara group to prevent complications of diarrhea and steatosis post LT[62].

PEBD: Partial external biliary diversion; PIBD: Partial internal biliary diversion; PFIC: Progressive Familial Intrahepatic Cholestasis; IE: Ileal Exclusion; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; TBD: Total biliary diversion; LT: Liver transplantation; NLS: Native Liver Survival; BSEP: Bile salt exporter pump; ESLD: End stage liver disease.
Antibody induced BSEP deficiency:Post-transplant recurrence of cholestasis in PFIC2 has been described due tode novopolyclonal antibodies directed against extracellular loop 1 domain of BSEP protein in the sera and on the canalicular membrane[63].Patients with severe mutations with resultant complete absence of BSEP are typically prone to this complication as the BSEP of the transplanted liver is recognized as a foreign new antigen by the recipient’s immune system, usually seen after an episode of acute rejection.Initial treatment of antibody-induced BSEP-deficiency disease is increased immunosuppression followed by either rituximab, bortezomib, intravenous immunoglobulin or plasmapheresis[64].Those refractory to the above treatment may be considered for allogeneic hematopoietic stem cell transplantation[65].Re-transplantmay be considered in those refractory to all therapy but the threat of recurrence in the re-transplanted liver is high[63].
Donor issues in living-related liver transplant:There have been apprehensions about using family members as donors, who may be heterozygous.Bassaset al[66] described living-related donor LT in 13 children with PFIC with good outcomes and no recurrence of symptoms in the recipients.Other studies have also reported favourable outcomes in PFIC2 and PFIC3 with living-related LT[67].Genetically proven heterozygous family members have been used as donors without recurrence of symptoms in the recipients.Currently, there is not enough evidence to perform a genetic analysis of the related donor before LT.It remains to be seen if the heterozygous donor liver in PFIC3 patients may predispose the recipient to drug-induced cholestasis, contraceptive induced cholestasis, ICP or GBD-1/LPAC.
Gene therapy:The additional complications associated with LT warrant the consideration of innovative therapeutic modalities like gene therapy.Recent approval for Adenovirus Associated viral (AAV) vector-based gene therapy for other monogenic disorders, raises hope for such future therapies for PFIC[68].The liver tropism of AAV vectors is a major advantage in the treatment of PFIC as this would imply the need for a lower vector dose for therapeutic efficacy.Higher AAV vector doses have been shown to cause adverse effects ranging from transient liver inflammation to even progressive liver failure and deaths in few reports[69].Gene therapy can be nonintegrating where each cell division post therapy will lead to some loss of episomal AAV vector genome or integratingin vivowhere these integrated genes are copied during cell division and passed on to the daughter cells without any loss of vector genome upon cell division[68].The benefit of integrating gene therapy is that a limited number of corrected hepatocytes will repopulate the liver and correct the defect[68,70].Theoretically,in vivonon-integrating AAV mediated gene therapy can be successful in PFIC3 as establishing ABCB4 expression in a proportion of hepatocytes.This can partially correct phosphatidylcholine transport and prevent the progression of the disease.Gene therapy for PFIC has only been done successfully in double knockout murine models (Abcb4-/-) to date[71].Treatment with modified mRNA variants encoding human ABCB4 encapsulated in lipid nanoparticles lead tode novoexpression of functional ABCB4 and restored phospholipid transport in cultured cells as well as Abcb4-/-mice.Hepatocyte damage due to accumulated BA in PFIC1 and 2; and decreased membrane stability in PFIC1 would warrant the correction of the genetic defect in all the hepatocytes, thus needing an integrating gene therapy strategy[68].Tumor genes is a possible major risk factor with integrating gene therapy as seen in trials on patients with severe combined immunodeficiency[72].The evolution of CRISPR/Cas9 based gene-editing tools that exhibit fewer off-target effects raises hope for integratingin vivocontrolled gene therapy in future[73].
Newer targets for therapy in PFIC: Several drugs and therapeutic molecules are currently being investigated for their therapeutic efficacy and safety in PFIC1 and 2.Table 4 and Figure 5 describes the various experimental drugs being evaluated for their therapeutic role in PFIC.Among them, the class of drugs that have generated the maximum interest are those which inhibit the ileal ASBTs.
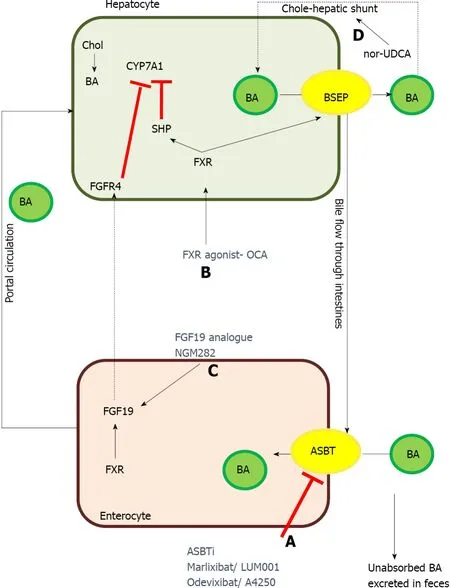
Figure 5 Therapeutic targets of some of the newer drugs for treatment of progressive familial intrahepatic cholestasis.
Apical sodium bile acid transporter inhibitors: ASBTi is currently under various stages of clinical trials include maralixibat and odevixibat (A4250).Currently, there are 4 registered clinical trials for maralixibat including one in infants and two clinical trials for odevixibat.The ASBTi interfere with bile salts reabsorption in the terminal ileum and hence greatly reduce the enterohepatic circulation of bile salts which are then excreted in the stool.ASBTi thus shrinks the bile acid pool and helps reduce BA and in turn improves cholestasis[74].Odevixibat is as effective as PEBD in reducing BA as well as pruritus in a child with PFIC2 thus raising hope of a very effective medical management of cholestasis[75].However, on stopping therapy after the completion of the trial, the patient had a relapse of symptoms and underwent PEBD.The preliminary results from the trials of maralixibat and odevixibat have shown that these ASBTi reduce BA and pruritus while improving growth[74,75].
Ivacaftor:Ivacaftor (VX-770), a CFTR/ABCC7 potentiator has been demonstrated to cause functional rescue of missense mutations of ABCB11 and ABCB4 located in a highly conserved ABC transporter motif[76].In vitromolecular modelling showed that ivacaftor led to the functional rescue of mutant ABCB4 resulting in increased phospholipid secretion[76].
4 phenyl butyrate:4-phenyl butyrate (4-PB), an FDA approved drug for urea cycle defect acts by 2 mechanisms to correct the defect in BSEP missense mutations: (1) It modulates the short-chain ubiquitination to prolong the degradation rate of cell surface resident BSEP and hence increases the cell surface expression of BSEP; and (2) Functions as a chemical chaperone to correct the misfolding of some endoplasmic reticulum-retained BSEP missense mutants[77].In vitrostudies in HEK293 and MDCKII cell lines showed increased cell surface expression of wild-type E297G and D482G BSEP.This resulted in an increased taurocholic acid transport on treatment with 4-PB, long term reduction in BA, improved presence of BSEP at canalicular membrane and relief of pruritus.4-PB therapy was effective at a dose of 500 mg/kg/d with no adverse events but with relapse of symptoms after stopping therapy[78].However, concerns have been raised about the palatability of this drug, compliance and hepatotoxicity potential when combined with rifampicin.Glycerol phenylbutyrate was shown to be equally efficacious with good tolerance and adherence.Malatacket al[79] described a case report of a PFIC2 with a missense mutation in BSEP who improved with combination therapy of 4-PB, maralixibat and oxcarbazepine.They hypothesized that enzyme inducer action of oxcarbazepine augments conversion of 4-PB to phenylacetate leading to synergistic effect apart from its usual nerve stabilizing effect[79].Although 4-PB is safe in most studies, a recent report of bipolar disorder in an adolescent on 4-PB raises concerns[80].
Readthrough therapy-gentamicin:Gentamicin has been shown to induce readthrough leading to the expression of a full-length protein in stable MDCK clones.Gentamicin in combination with 4-PB significantly increased the readthrough level of common nonsense mutation studied (p.R415X, p.R470X, p.R1057X, p.R1090X) in HEK293 cells.The maximum response was seen for the p.R1090X mutation with a 40% increase in taurocholate transport and correct localization at the cell membrane[81].There has been an emphasis on mutation-specific therapies with: (1) Gentamicin and ataluren likely to suppress nonsense mutations by promoting the readthrough of premature stop codons; and (2) U1-small nuclear-RNA likely to rescue splicing for several ATP8B1 mutations located at donor, acceptor and splice sites[81].
Other drugs:Nor-UDCA is a side-chain shortened derivative of UDCA with relative resistance to amidation thus increasing the cholehepatic shunt and in turn increases the bile salt-dependent bile flow[82].Peroxisome proliferator-activated receptor agonist bezafibrate has been reported to correct dyslipidemia and reduce pruritus and cholestasis in children with PFIC1 and other diseases[83].Rapamycin restores bile acid excretion, attenuates hepatocyte damage and extends the lifespan of Abcb11b mutant zebrafish which is the ortholog of human ABCB11.Other drug categories which could theoretically be beneficial but have not yet been tried in clinical trials include FXR agonist and fibroblast growth factor-19 analogue[84].Obeticholic acid is a very potent FXR agonist, promotes transcription of BSEP, improves histopathology and reverse fibrosis and could therefore be useful in patients with PFIC2[84].
CONCLUSION
To conclude, there has been a recent emphasis on understanding the genetic spectrum of PFIC, their phenotype, natural course and long-term outcome.Genotype correlates well with phenotype in PFIC2 but not in PFIC1.The expanded role of the heterozygous transporter defects presenting late needs to be suspected and identified even in adulthood.Medical therapy and SBD form the cornerstone of the management of pruritus.LT in these children is associated with unique issues like a high rate of intractable diarrhea, growth failure, steatohepatitis and graft failure in PFIC1 and antibody-mediated BSEP deficiency.There is a promising role of ASBT inhibitors in the management of cholestasis.
杂志排行

World Journal of Hepatology的其它文章
- Hepatitis C virus: A critical approach to who really needs treatment
- Current aspects of renal dysfunction after liver transplantation
- Hepatitis C: Problems to extinction and residual hepatic and extrahepatic lesions after sustained virological response
- Metabolic and nutritional triggers associated with increased risk of liver complications in SARS-CoV-2
- Targets of immunotherapy for hepatocellular carcinoma: An update
- Redefining non-alcoholic fatty liver disease to metabolic associated fatty liver disease: Is this plausible?