Stearoyl-CoA desaturase 1: A potential target for non-alcoholic fatty liver disease?-perspective on emerging experimental evidence
2022-02-12ShanmugamMurugaihaJeyakumarAyyalasomayajulaVajreswari
Shanmugam Murugaiha Jeyakumar, Ayyalasomayajula Vajreswari
Shanmugam Murugaiha Jeyakumar, Ayyalasomayajula Vajreswari, Division of Lipid Biochemistry, National Institute of Nutrition, Hyderabad 500007, Telangana, India
Shanmugam Murugaiha Jeyakumar, Department of Clinical Pharmacology, National Institute for Research in Tuberculosis, Chennai 600031, Tamil Nadu, India
Abstract Non-alcoholic fatty liver disease (NAFLD) is a progressive disease and one of the leading causes of death.An unnamed disease has become a global epidemic disease of public health concern.This spectrum of diseases manifests itself with initial accumulation of excessive triglycerides (due to de novo lipogenesis) in the hepatocytes, leading to simple steatosis.Although its aetiology is multi-factorial, lifestyle changes (diet and physical activity) are considered to be the key thriving factors.In this context, high fructose consumption is associated with an increased risk for developing NAFLD in humans, while high-fructose feeding to experimental animals results in hepatic steatosis and non-alcoholic steatohepatitis, by increasing hepatic lipogenesis.Among several lipogenic genes, the endoplasmic reticulum-bound stearoyl-CoA desaturase 1 (SCD1) is the key determinant of triglycerides biosynthesis pathway, by providing monounsaturated fatty acids, through the incorporation of a double bond at the delta-9 position of saturated fatty acids, specifically, palmitic (C16:0) and stearic (C18:0) acids, yielding palmitoleic (C16:1) and oleic (C18:1) acids, respectively.Various experimental studies involving SCD1 gene knockout and diet-induced rodent models have demonstrated that SCD1 plays a key role in the development of NAFLD, by modulating hepatic lipogenesis and thus triglyceride accumulation in the liver.Several pharmacological and dietary intervention studies have shown the benefits of inhibiting hepatic SCD1 in the pathogenesis of NAFLD.In this review, we give an overview of SCD1 in NAFLD, based on the current experimental evidence and the translational applicability of SCD1 inhibition in human NAFLD conditions, besides discussing the limitations and way-forward.
Key Words: Steatosis; Lipids; Fatty acids; Vitamin A; Metabolic syndrome; Obesity
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is a spectrum of diseases including simple hepatic triglyceride accumulation, otherwise called steatosis, to hepatocellular carcinoma (HCC).Although its aetiology is multi-factorial, lifestyle modifications and genetic susceptibility are considered as the major thriving forces for the development of NAFLD, besides obesity and metabolic syndrome.Like other metabolic diseases, NALFD also contributes to the development of insulin resistance, metabolic syndrome, and obesity.Various external and internal factors influence its progression from simple steatosis to the end-stage disease HCC, which takes several years with an incidence rate ranging between 2.4% to 12.8%.As the aetiology and progression of NAFLD are asymptomatic, diagnosis, control, and management of NAFLD at an early stage are very much challenging.Further, so far no specific therapy to treat the NAFLD has been identified, except weight management therapy[1,2].
Studies from several genetically-engineered rodent models have demonstrated the involvement of numerous genes in the development and/or progression of NAFLD, namely, adiponutrin/patatin-like phospholipase domain-containing protein 3 (PNPLA3), caspases 1 and 3, cannabinoid receptor 1, hepcidin, prolyl endopeptidase, stearoyl-CoA desaturase 1 (SCD1), and thyroid hormone receptor-α, to name a few[3-10].Besides, Coleet al[11] have described various genetic, drug-induced, and other NAFLD models for drug discovery and their potential use in therapeutics.Previously, Postic and Girard[12] have detailed the role of several genes that are involved in the hepaticde novolipogenesis and their interaction with fatty acid oxidation, triglyceride secretion, and thus hepatic steatosis and its associated complications, including insulin resistance, based on the studies from the genetically engineered mice.In this review, we primarily focus on the role of SCD1 in the development of hepatic steatosis from various experimental models and discuss the potential scope of its inhibition in ameliorating NAFLD, besides highlighting the limitation, especially, the existing translational research gap between the experimental research and its extension to clinical research in the control and/or management of NAFLD.
NAFLD: AN UNNAMED DISEASE TO A GLOBAL EPIDEMIC DISEASE
NAFLD is a spectrum of several related diseases in the absence of alcohol consumption as the etiological origin.The earliest stage in NAFLD is hepatic steatosis/fatty liver, which is characterized by the deposition of triglycerides in the cytoplasmic lipid droplets of hepatocytes.The hepatic steatosis/fatty liver is often selflimiting; however, it can progress to non-alcoholic steatohepatitis (NASH), the condition characterized by the presence of hepatocyte injury (hepatocyte ballooning and cell death), infiltration of immune cells, inflammatory mediators, and activated stellate cells.Due to the vicious cycle of inflammatory insults and stellate cell activation, NASH progresses to fibrosis and cirrhosis, which can eventually progress to HCC, thus resulting in hepatocellular death[1,2].
In the year 1952, Zelman[13] has reported liver damage in obese humans based on liver function tests and liver biopsy examination.In 1958, Westwater and Fainer[14] have confirmed liver damage in obese patients, as evidenced by abnormal liver function and histology.Adler and Schaffner[15], who have examined a group of 25 overweight patients for the presence of fatty liver, fatty hepatitis, fatty fibrosis, and cirrhosis, based on liver biopsy and function, have reported that an equal frequency of all these pathological conditions.Further, these hepatic pathological changes resembled the liver damage caused by alcohol and post-jejuno-ileal bypass surgery.Ludwiget al[16], who have studied liver disease in 20 obese patients in Mayo Clinic, have found similarities between the hepatitis of unknown cause and the alcoholinduced hepatitis with respect to the histological changes, such as fatty changes, lobular hepatitis, and focal necrosis with mixed inflammation.Further, they have coined the term NASH in 1980 for the first time; until then it is known as an unnamed liver disease.The current global prevalence of NAFLD is estimated to be 24%, which has increased from 15% to 25% between 2005 and 2010, and the data from a recent meta-analysis study on the general population have shown a higher prevalence of NAFLD in the Middle East (32%) and South America (31%), followed by United States (24%), while being the lowest in Africa (14%)[17,18].Further, in Asia, the overall prevalence of NAFLD is estimated to be 29.6%[19].Undoubtedly, NAFLD is now a global epidemic disease of public health concern and therefore, its control and management are the top research priorities.Further, along with other noncommunicable diseases that include obesity, type 2 diabetes, and metabolic syndrome, it contributes to the global disease burden and associated health and economic consequences.
AETIOLOGY AND PATHOGENESIS OF NAFLD
The natural history of NAFLD in terms of its occurrence or causation and pathogenesis is multi-factorial, poorly understood, and further complicated by the involvement of the host’s genetics and interactions with lifestyle changes including various environmental factors and other pre-existing co-morbidities and risk factors.Nevertheless, some of the key underlying mechanisms involved in the hepatic triglyceride accumulation are increased hepaticde novolipogenesis, diminished export of triglycerides through lipoproteins, and impaired β-oxidation of free fatty acids[12].However, the pathogenesis/progression of NAFLD, from fatty liver to hepatocellular death, is explained initially by the two-hit hypothesis.Subsequently, it is substituted by the parallel, multiple-hit hypothesis.Accordingly, the first insult is initiated by the accumulation of lipids, particularly, triglycerides inside the hepatocytes and the development of hepatic insulin resistance.This causes the activation of several cascades of events both at hepatic and extra-hepatic sites, particularly adipose tissue that ultimately leads to the excessive free fatty acid influx, increased lipogenesis, and triglyceride accumulation.Parallelly, this causes a perpetual cycle of multiple insults to the hepatocytes through cellular stress (oxidative and endoplasmic reticulum stress), mitochondrial dysfunction, dysbiosis, inflammatory response, and hypoxia, to name a few and mediated by the interplay between several cell types of hepatic, extrahepatic and systemic origins[20,21].Although certain pharmacological agents (lipidlowering drugs, such as metformin and statins) and weight management therapy are offered, there are no specific drugs to treat NAFLD (except managing the disease conditions) due to high complexity and poor understanding of its pathogenesis[22].
DIETARY CHANGES AND NAFLD
Sugars are naturally occurring sweeteners, and sucrose, fructose, and glucose are the most common sugars in our daily diet.Before the industrial era, the amount of fructose was very low in the human diet and derived mainly from natural resources such as honey, dates, raisins, grapes, raw apples, squeezed apples, persimmons blueberries, and molasses.After industrialization, sweeteners are produced on a large scale from various sources, particularly corn.During this process, starch isolated from corn, is initially hydrolyzed into glucose, and followed by the enzymatic isomerization of the released glucose into fructose.The resultant product/mixture is known as highfructose corn syrup (HFCS).Relative to sucrose, the usage of HFCS in the food industries is high, due to its low cost and sweeter taste and also as it stabilizes the texture of processed food better than sucrose.The most widely recognized type of HFCS is HFCS 55, having 55% fructose compared to sucrose which has 50% fructose[23-25].Although both glucose and fructose are simple carbohydrates, unlike glucose, the absorption and metabolism of fructose are completely different.Moreover, it is more lipogenic than glucose.Therefore, excessive consumption of fructose causes uncontrolled lipogenesis and triglyceride synthesis in the liver, due to the lack of ratelimiting enzyme or metabolic check-point[26].Hepaticde novolipogenesis is considered to be an important contributing factor in the development of NAFLD[27].Donnellyet al[28] have shown that in the fasted state, 26% of triglyceride and 23% of very-low-density lipoprotein (VLDL)-triglyceride in the liver of NAFLD patients are derived from thede novolipogenesis.In addition, Lambertet al[29] have shown that, compared to the control subjects,de novolipogenesis is 3% higher in the NAFLD subjects.Although contradictory findings exist, most of the epidemiological and clinical studies have shown the association between high fructose consumption (majorly in the form of HFCS) and the risk of NAFLD causation and other metabolic complications, including obesity, insulin resistance, and metabolic syndrome[30-34].
SCD1-A REGULATOR OF LlPOGENESlS
SCD1 is an endoplasmic reticulum-bound microsomal enzyme that catalyses the formation of monounsaturated fatty acids (MUFA) from saturated fatty acids (SFA) by incorporating a double bond at the delta-9 position, by involving cytochrome b5, NADPH-dependant cytochrome b5 reductase, and molecular oxygen.Palmitoleic (C16:1) and oleic (C18:1) acids are the SCD1-catalyzed products from their respective substrates palmitic (C16:0) and stearic (C18:0) acids[35,36].SCD1 is abundantly expressed in adipose tissue and the liver, though different isoforms of SCD have been identified in various species including humans, such as SCD 1-4 in mice, SCD1 and 2 in rats, and SCD1 and 5 in humans.These isoforms display differential expression pattern and tissue specificity, however, the role of some of these isoforms is not fully elucidated.As constituents of cell membranes, MUFA play a crucial role in maintaining membrane fluidity.Therefore, the altered ratio of SFA to MUFA in membranes affects the fluidity, thereby modulating the cellular signalling and physiological functions[36].
SCD1 is the rate-liming enzyme of synthesis of MUFA, which are the major substrates for the synthesis of triglycerides, phospholipids, and cholesteryl and wax esters.Diet-derived and the endogenously (fatty acid biosynthetic pathway) formed palmitic acid (C16:0) and its chain elongation product stearic acid (C18:0) are desaturated by the SCD1 and the newly formed MUFA,i.e.,palmitoleic (C16:1) and oleic (C18:1) acids, respectively, are preferably esterified with glycerol-3-phosphate to form lysophosphatidic acid, the first step of triglyceride assembly by the enzyme glycerol-3-phosphate acyltransferase (GPAT).After several enzymatic steps, finally, it results in the formation of triglycerides by the action of diacylglycerol acyltransferase (DGAT) and it is either stored in the liver or assembled into VLDL and exported to extra-hepatic tissues[37].The SCD is a critical metabolic control enzyme, as its activity determines the fate of fatty acids by diverting them to either oxidation or storage, and hence, modulates the energy homeostasis and thereby obesity.This is evident from theSCD1gene knock-out mouse study of Ntambiet al[38].Earlier, a study from our lab has shown that fatty acid desaturation indices (the ratio of product to the substrate;i.e.C16:1/C16:0 and C18:1/C18:0) of SCD 1 are associated with body mass index and adiposity in genetically obese rat models[39].The dysregulated SCD1 is considered to be one of the key mediators in the pathophysiology of several metabolic and/or inflammatory diseases, including obesity, metabolic syndrome, diabetes, NAFLD, cardiovascular diseases, and cancer[40-43].Importantly, SCD1 is regulated by numerous nutritional (fatty acids, cholesterol, vitamin A, and iron) and hormonal (leptin and thyroid hormone) factors[44-49].
SCD1 AND NAFLD - EXPERlMENTAL EVlDENCE
The very first time, from gene-knockout mouse models (SCD1-/- andSREBP1c-/-), Miyazaki and colleagues have reported that SCD1 and its enzymatic product oleate (C18:1) are essential for fructose-induced hepatic lipogenesis and triglyceride synthesis through both sterol regulatory element-binding protein 1c (SREBP-1c)-dependent and independent pathways[50].A study based on globalSCD1knock-out mice has demonstrated that SCD1 deficiency resulted in the increased expression of genes involved in the fatty acid oxidation, while decreased the key lipogenic genes, thereby decreasing the triglyceride synthesis and secretion by the liver.Further,SCD1gene knockout with leptin deficiency,i.e.,inob/obmice, has resulted in the attenuation of the hepatic triglyceride accumulation and secretion of VLDL.It has been reported thatSCD1gene knock-out mice display increased hepatic mitochondrial fatty acid oxidation, which is evident from the increased activities of carnitine palmitoyltransferase (CPT), the gate-keeper enzyme of β-oxidation.Further, the authors have reported that the effects are mediated through the activation of the adenosine monophosphate (AMP)-activated protein kinase (a metabolic sensor) due to the deficiency of SCD1.Further, the SCD1 mutation has also led to AMPK activation inob/obmice[51].Miyazakiet al[52] have shown in a natural homozygousSCD1gene mutated asebia mouse model that the absence of SCDI has led to the impaired hepatic synthesis of cholesterol ester and triglycerides.In a liver-specificSCD1knock-out mouse model, Miyazakiet al[9] have found that these mice are resistant to highcarbohydrate (high sucrose and very low-fat) diet-induced adiposity and hepatic steatosis.In a genetically modified NAFLD mouse model that possesses Nglycosylated cyclic AMP-responsive element-binding protein H (CREBH) (endoplasmic reticulum-anchored transcription factor), there was decreased production of peroxisome proliferator-activated receptor α (PPARα) and activity of SCD1, which in turn resulted in the reduction of hepatic lipid accumulation, lipotoxicity, and inflammation[53].The study of Flowerset al[54] has shown that SCD1 deficiency has diverse effects on lean and obese mice, as evidenced by improved insulin sensitivity in the former, while aggravation of diabetes (due to pancreatic beta-cell loss) in the latter.
Jianget al[55] have demonstrated that the pharmacological inhibition of SCD1 through the anti-sense oligonucleotide has resulted in increased fatty acid oxidation and reducedde novofatty acid synthesis and thus steatosis both in hepatocyte cell line and mouse models.Non-coding ribonucleic acids microRNA-103, 212-5p, and 27a have been shown to suppress the SCD1 in the liver, besides fatty acid synthase (FAS), and thus reduced the diet-induced obesity, hepaticde novolipogenesis, and hepatic lipid accumulation as evidenced byin vivoandin vitromodels[56-58].Oral administration of a novel SCD-1 inhibitor, N-(2-hydroxy-2-phenylethyl)-6-[4-(2-methylbenzoyl) piperidin-1-yl] pyridazine-3-carboxamide, has been shown to attenuate hepatic lipid accumulation and histological features of NASH, such as hepatocellular degeneration, inflammation, and liver injury in an NASH rat model[59].Another study using an SCD1 selective inhibitor, 3-[4-(2-chloro-5-fluorophenoxy)-1-piperidinyl]-6-(5-methyl-1,3,4-oxadiazol-2-yl)-pyridazine, has shown a reduction in triglyceride accumulation and promoted liver-specific functions, during the multiple stages of hepatocyte differentiation in human pluripotent stem cells.Further, the authors have observed the MUFA oleate-mediated reversal of SCD1 inhibition.In addition, the authors could find some of these changes due to SCD1 inhibition, during differentiation, in human primary mononuclear cells (hPMN)[60].Iida and colleagues[61] have discovered a synthetic compound, thiazole-4-acetic acid analogue 48, displaying liver-specific inhibition of SCD1.Further, the investigators have demonstrated the pharmacological effects (such as anti-diabetic and anti-obesity) of hepatic SCD1 inhibition in rodent models of metabolic diseases such as diabetes, obesity, and hepatic steatosis using this analogue.In addition, pre-clinical toxicological evaluation of this compound has displayed no significant adverse events and therefore, the authors have concluded that the compound has a potential therapeutic utility in treating some of the chronic diseases[61].In Zucker fatty rats (fa/fa), oral administration of an SCD1 inhibitor, GSK993, decreased the hepatic lipids, and improved impaired glucose tolerance and insulin sensitivity[62].Taoet al[63] have shown that the intraperitoneal administration of α2-adrenoceptor agonist dexmedetomidine (DEX) to diet-induced NAFLD mice resulted in the inhibition of hepatic steatosis and improvement of insulin sensitivity and inflammation, associated with a significant reduction in hepatic SCD1 mRNA and protein levels[63].Previously, Attieet al[64] who have assessed the SCD1 and its association with plasma triglycerides have reported an association among hepatic SCD1 activity, fatty acid desaturation index (the ratio of C18:1/C18:0), and plasma triglyceride levels in mice, while a twofold elevation of desaturation index is associated with a four-fold increase in plasma triglyceride concentration in humans.It has been shown that the SCD1 fatty acid desaturation index (i.e.C16:1/C16:0) correlates with fatty liver index of dyslipidaemic individuals, and importantly, the total PUFA were inversely associated with SCD1, thus NAFLD[65].Qinet al[66] have shown that SCD1-mediated lipid desaturation plays a critical role in HCC, by modulating endoplasmic reticulum (ER) stress.Zhouetal[67] who have studied the underlying mechanism of the development of hepatic steatosis have reported that the AMPK activation and lipophagy are the key mediators of SCD1 inhibition-induced amelioration of fatty liver as demonstrated in primary hepatocytes and high-fat diet-fed mice.
Quality and quantity of lipids/fats are known to alter the expression of SCD1 in mice susceptible to diet-induced metabolic diseases, including atherosclerosis, diabetes, obesity, and certain types of cancers[68].In a study of Sekiyaet al[69], dietary PUFA-fedob/obmice displayed SREBP-1-mediated suppression of lipogenic genes, includingSCD1, and thus reduced hepatic triglyceride contents and liver enzymes, in addition to hyperinsulinemia and hyperglycemia.Mark Brownet al[70], who have investigated SCD1 inhibition on metabolic syndrome and atherosclerosis in experimental rat models, found that the SCD1 inhibition protected the mice from developing metabolic syndrome and prevented atherosclerosis synergistically by the treatment with fish oil and anti-sense oligonucleotide-targeted SCD1 suppression[70].MacDonaldet al[71] reported that decreased SCD1 activity is associated with improved metabolic syndrome phenotypes, including the reduction in plasma triglycerides, non-high-density lipoprotein (HDL) cholesterol, VLDL triglycerides, hepatic steatosis, fat mass, and insulin resistance induced by a Western diet in a low-density lipoprotein receptor-deficient mouse model.Conjugated linoleic acid (CLA) isomers have been shown to attenuate fructose-induced hepatic lipogenesis, lipid accumulation, and hypertriglyceridemia, through the suppression of lipogenic genes SCD1 and FAS of the liver[72].Zhuet al[73] have reported that metformin, an anti-diabetic drug, ameliorates triglyceride accumulation by inhibiting hepatic SCD1 in the HepG2 cell line.Earlier, a study from our lab showed that SCD1 is a key player in fructoseinduced hepatic triglyceride accumulation.However, for the first time, it has been demonstrated that a high fructose diet sans vitamin A failed to induce hepatic steatosis, while replenishment with vitamin A restored the fructose-induced triglyceride accumulation, suggesting that vitamin A is essential for fructose-induced metabolic alterations in the liver, associated with triglyceride metabolism[74].Overall, targeting the SCD1 by knocking out the gene, dietary factors, and chemical inhibitor results in the reduction of its mRNA or protein or activity and MUFA levels.These events have been associated with improved NAFLD and/or its associated complications eventually, which include hepatic steatosis, NASH, liver injury, hepatocellular degeneration, hypertriglyceridemia, inflammation, hyperinsulinemia, impaired glucose tolerance, insulin sensitivity, hyperglycemia, and obesity.Notably, most of these metabolic complications are characteristic features of NAFLD in humans as well.Therefore, SCDI has significant clinical implications and apparently, SCD1 is a potential target for treating NAFLD in humans.Nevertheless, there are limitations to achieve the translational potential of SCD1 inhibition in clinical situations (Schematic summary is given in Figure 1).
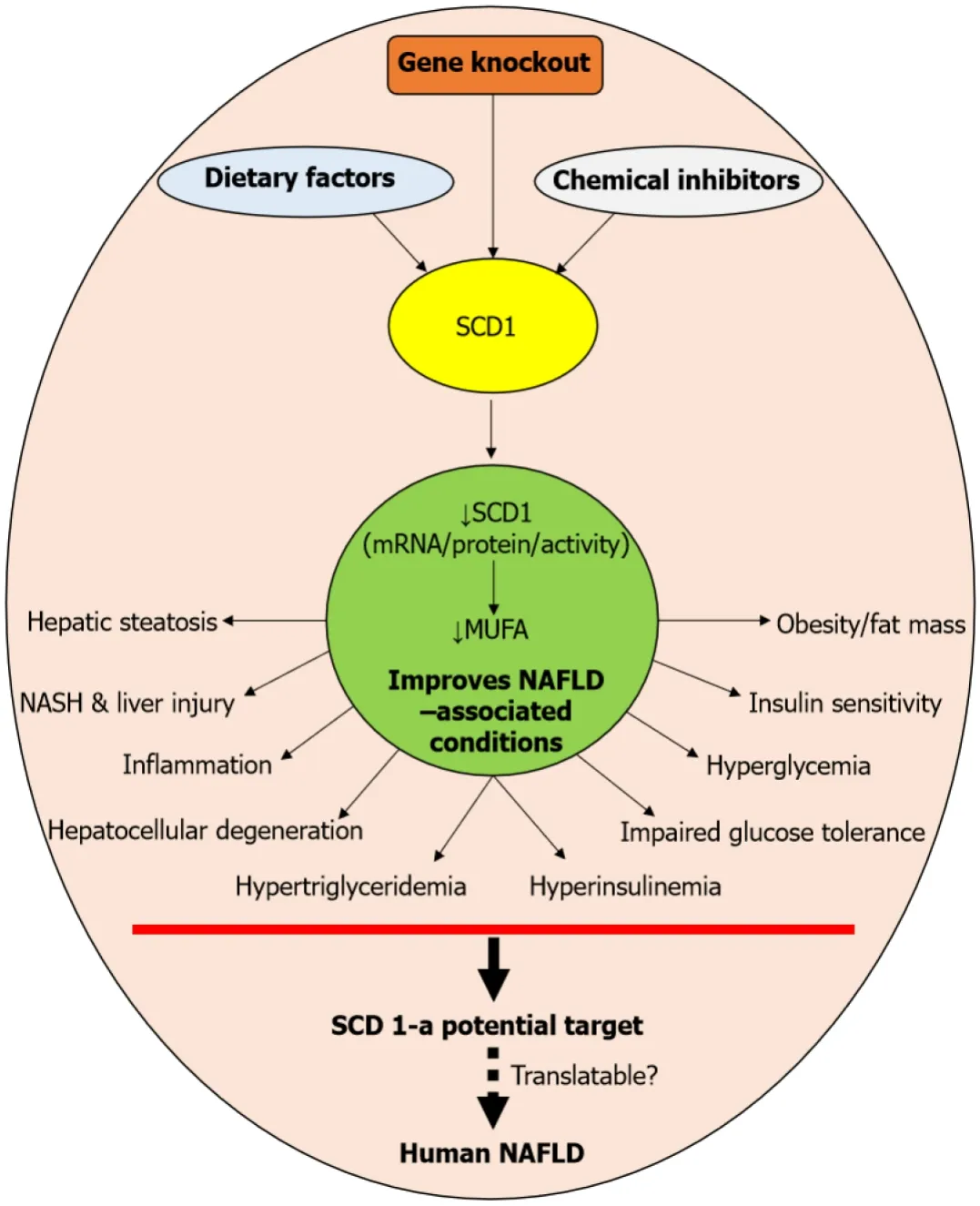
Figure 1 Schematic summary of experimental evidence on stearoyl-CoA desaturase 1 inhibition.
TARGETlNG SCD1 lS FAR FROM TRANSLATlONAL APPLlCABlLlTY? - CHALLENGES AND WAY-FORWARD
So far experimental evidence from genetic, diet-induced rodent models as well as from supplementation and interventional studies has demonstrated that SCD1 is the central player in lipid metabolism, energy homeostasis, and thus obesity and NAFLD.This has given enormous hope for its clinical utility and driven various pharmaceutical companies to develop potent inhibitors for SCD1.Earlier, Powell[75] has over-viewed several small molecule SCD1 inhibitors (such as piperazinyl pyridazine-based derivatives/analogues, cyclic urea, spirocyclic compounds, bicyclic heteroaromatics, triazole and aryl/heteroaryl linkers, piperidine aryl ketones, aryl diamine, bicyclic aryl diamine linkers, pyrazole and triazole derivatives/analogues, pyridazine-2-one and triazine derivatives to name a few of them) that are patented by pharmaceutical companies during 2009 to 2013 and their potential application in various metabolic diseases, such as obesity, diabetes and cancer.Further, the author has underlined the fact that the safety and efficacy of these inhibitors in humans remain unanswered.Recently, Uto[76] has articulated the current advances in the area of SCD1 inhibitor development and highlighted some of the tissue- or disease-specific SCD1 inhibitors.However, the author has also pointed out the knowledge gap in understanding the role of SCD1 in humans, in addition to the therapeutic applications of these inhibitors in clinical settings[76].
Unlike liver-specific inhibition, global SCD1 inhibition or deficiency displays detrimental effect on various organs, particularly, the skin and eyes, and these aspects have been extensively reviewed earlier by Zhanget al[77].However, a comprehensive understanding of other metabolic changes or distortion and the susceptibility to other metabolic insults or dietary and environmental factors due to liver-specific SCD1 inhibition is not even at the experimental stage.It has been shown that hepatic SCD1-deficient mice are susceptible to chemically-induced ulcerative colitis, besides resulting in the elevation of pro-inflammatory responses[78].Aljohaniet al[79] have reported that liver-specific SCD1 deficiency increases ER stress by activating the mammalian target of rapamycin complex 1 (mTORC1) in the globalSCD1knockout mouse model; however, oleate has been shown to deactivate the mTORC1 signalling and dissolute ER stress.SCD1-mediated ER stress in HCC through lipid desaturation has also been reported[66].Buschet al[80] have shown that increased SCD1 and its fatty acid desaturation index have a protective effect on SFA; palmitate-induced pancreatic beta-cell apoptosis and inhibition of SCD1 by CLA have also offered protection against lipotoxic effects of the palmitate.In line with this, previously, a study from our lab has also shown that the suppression of SCD1 and thus the MUFA oleic acid (C18:1) is associated with increased ER stress in the pancreas and hence islet cell apoptosis and decreased pancreatic hormones, namely insulin, glucagon, and Cpeptide[81].Notably, in one of our studies, we have reported that despite a reduction in the liver SCD1, there is no improvement in high fructose diet-induced hepatic steatosis[82].Therefore, the inhibition of SCD1 may not lead to an improvement in hepatic steatosis, at least in certain conditions.
Since NAFLD is a benign and asymptomatic disease, identifying or diagnosing it at an early stage is very challenging.Importantly, there are no reliable and specific circulatory markers to identify the occurrence and/or classify the stages of NAFLD.Notably, Yamadaet al[83], who have analysed the liver fatty acid composition and gene expression in patients with NASH, have reported the prevailing differences in these parameters among patients with simple steatosis and NASH.In another important study, Teufelet al[84] have reported the significant differences in the expression pattern of several pathway genes associated with NAFLD/NASH between murine models and human liver tissue, along with substantial differences in the pathogenesis of NAFLD between these two species.So far, the available data have demonstrated the modulatory effect of SCD1 on the initial stage of NAFLD development, particularly, hepatic steatosis and NASH, which are largely derived from experimental studies.Therefore, there is much ambiguity with regard to the inhibition of SCD1, whether it will retard/arrest the progression and/or reverse the conditions of NASH and subsequent stages of NAFLD in humans.Furthermore, the regulatory role of SCD1 in different stages of NAFLD (fibrosis, cirrhosis, and HCC) is poorly understood even in experimental models and more so in humans.In addition, unlike other developed and developing countries, in India, a higher proportion of NAFLD has been reported in lean subjects, whose BMI is < 23 kg/m2[85].However, the role of SCD1 in lean NAFLD has not been addressed or defined adequately so far.Leeet al[86] have reported sex-specific differential expression of hepatic SCD1 in mice.More importantly, in the recent past, the sexual-dimorphic pathophysiology of NAFLD in humans has also been well received[87-89].Unlike in rodents, the functions of SCD1 and SCD5 in humans are not well characterized and fully understood.In the NAFLD spectrum, besides the liver, several other players modulate the development and pathogenesis of NAFLD, and particularly the adipose tissue (which abundantly expresses SCD1), through a wide range of secretory adipocytokines[90].Emerging evidence suggests that the pathogenesis of NAFLD involves an interplay of multiple organs in a system, in addition to environmental factors[90,91].In such a case, it is unclear whether targeting/inhibiting the hepatic SCD1 alone would yield the desired clinical outcomes in NAFLD? Similarly, several questions are yet to be answered and the knowledge gaps need to be addressed in both experimental and clinical NAFLD.Hopefully, in the coming years, the technological advancements in the life sciences (omics, patient/human-derived organoids,etc.) and computational science (in silico, AI-based tissue modelling, and tools for prediction, diagnosis, and prognosis) would shed light on some of these grey areas.
CONCLUSION
The endoplasmic reticulum-bound SCD1 enzyme plays a very critical role in the development of NAFLD, by altering the hepatic MUFA concentration.The literature is replete with the reports demonstrating the role of SCD1 in the causation and pathogenesis of NAFLD.Notably, the liver-specific inhibition of SCD1 has been shown to attenuate the development of hepatic steatosis and thus NAFLD in several genetic and diet-induced experimental models, besides supplementation and intervention studies (diet and pharmacological agents).Although these experimental data are encouraging, the role and regulation of SCD1 in the human NAFLD conditions are poorly understood and thus need further research in this direction.Nevertheless, so far, the existing quantum of experimental and some supporting clinical data suggests that the SCD1 is a potential target and infuse a strong hope for translational applicability of SCD1 inhibitors, as a therapeutic option.Certainly, the inhibition of SCD1 would help in the control and/or management of NAFLD in humans.
杂志排行

World Journal of Hepatology的其它文章
- Hepatitis C virus: A critical approach to who really needs treatment
- Current aspects of renal dysfunction after liver transplantation
- Hepatitis C: Problems to extinction and residual hepatic and extrahepatic lesions after sustained virological response
- Metabolic and nutritional triggers associated with increased risk of liver complications in SARS-CoV-2
- Recent updates on progressive familial intrahepatic cholestasis types 1, 2 and 3: Outcome and therapeutic strategies
- Targets of immunotherapy for hepatocellular carcinoma: An update