臭氧催化氧化降解煤化工高盐废水有机物的机理
2022-02-12王吉坤李阳陈贵锋刘敏寇丽红王琦何毅聪
王吉坤,李阳,陈贵锋,刘敏,寇丽红,王琦,何毅聪
(1 煤炭科学技术研究院有限公司,北京 100013;2 煤炭资源高效开采与洁净利用国家重点实验室,北京 100013)
国家环保要求煤化工废水须“零排放”处理。现有煤化工废水处理工艺主要为“预处理+生化处理+深度处理”,其中深度处理工艺主要为多级膜浓缩和蒸发结晶工艺,经深度处理后产生了大量高盐废水,其中废水中的主要盐离子有Na、Cl、SO2等,废水中难降解有机物主要为腐殖质类、芳香族化合物等。高盐废水中难降解有机物导致蒸发结晶的杂盐率升高,杂盐须按危险废弃物处置,导致企业运行成本增加。因此有效去除难降解有机物是实现废水零排放的关键。
高盐废水降解有机物的方法众多,主要有光催化氧化法、电解法、芬顿氧化法及臭氧(O)催化氧化法等。其中光催化氧化法对废水水质色度要求高,催化降解效率较低,且运行成本高,工程应用少;电解法存在电极价格昂贵且电极寿命低的缺点,制约了电解法在高盐废水领域的推广使用;光芬顿氧化法在氧化过程中需不断调节pH,同时该过程易产生铁泥,增加出水盐含量及增加膜浓缩及蒸发结晶的负荷,一般不适于高盐废水难降解有机物的去除。
相比上述方法,臭氧催化氧化法通过产生氧化能力更强的·OH(羟基自由基)的方式去除高盐废水中的难降解有机物。诸如Pi等采用臭氧催化剂降解草酸废水,总有机碳去除率相比纯臭氧氧化法提高30%;Rosal 等采用臭氧催化剂降解非诺贝酸废水,羟基自由基生成速率是纯臭氧氧化的8 倍;王吉坤等采用浸渍法制备催化剂,开展臭氧催化氧化去除煤化工高盐废水工艺参数优化研究,确定了最佳工艺参数;马栋等研究了不同活性组分的臭氧催化剂对COD 去除率的影响,发现过渡金属元素(Cu、Mn、Ni、Fe、Co)对COD 去除率高于稀土元素(Ce);Wen 等制备锌系催化剂用以去除草酸及溴酸盐,发现30min后草酸去除率为70.9%,溴酸盐去除率为99.4%;Qiang 等采用铁系催化剂用以降解农药氧化乐果,发现在催化剂产生·OH,其与O反应速率常数为5.3×10m·s;Zhai 等制备纳米ZnO 催化剂去除水中二氯乙酸,发现催化剂比O降解二氯乙酸的去除率提高了32.5%;Lyu等采用铁钴系催化剂降解水中酸类及酚类有机物,发现钴铁系催化剂对有机物的去除率达到93%,比铁系催化剂的去除率提高了33%。综上所述,臭氧催化氧化法在煤化工高盐废水有机物去除上应用前景广阔。但目前臭氧催化氧化技术主要集中在催化剂性能评价及非高盐水的机理研究上,对高盐废水降解机理的研究尚未见相关文献报道,因此有必要开展臭氧催化氧化降解有机物的机理研究。
本文采用浸渍−焙烧法制备臭氧催化剂,开展煤化工高盐废水的臭氧催化氧化实验,确定最佳催化剂;对臭氧催化剂开展表征分析,了解催化剂的形貌特征、元素组成、电位特征;最后对臭氧催化氧化机理开展研究,明确催化剂降解有机物的作用机理及反应历程,以期为臭氧催化剂的制备及臭氧催化氧化降解有机物效率的提高提供理论指导。
1 材料和方法
1.1 实验药品
甲酸(分析纯)、硝酸铁(分析纯)、硝酸锰(分析纯)、硝酸铈(分析纯)、硝酸铜(分析纯)、硅铝球(3~5mm)。
1.2 实验仪器
紫外分光光度计、pH 计、COD 快速消解测定仪、生化培养箱、多参数水质测定仪、马弗炉、振荡器。
1.3 实验水样分析
实验水样来自国内三个典型煤化工企业,水样1 为内蒙古某煤制气高盐废水,水样2 为内蒙古某煤制甲醇高盐废水,水样3为河北某煤焦化高盐废水,对废水进行分析,结果见图1。
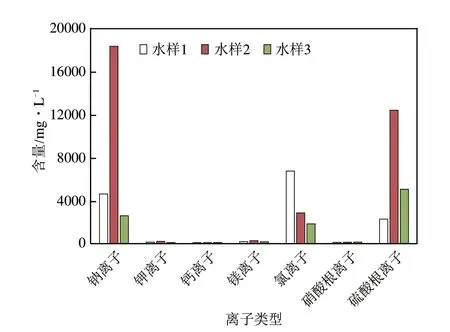
图1 高盐废水离子类型
由图1可知:三种煤化工高盐废水中阴阳离子种类及含量随工艺不同而各不相同,但三种煤化工高盐废水阳离子主要为钠离子,还含有少量的钾离子、钙离子及镁离子;水中阴离子主要为氯离子、硫酸根,其次为硝酸根离子,其中水样1中氯离子含量与硫酸根离子含量比约为3∶1,水样3中氯离子含量与硫酸根离子含量比约为1∶2。
1.4 臭氧催化剂载体选择
目前现有的催化剂载体有陶粒、活性氧化铝及硅铝球载体。因陶粒的主要成分为氧化硅,氧化硅没有酸位点,没有催化活性;活性氧化铝虽然表面上可以形成部分羟基,但也仅仅表现出很弱的B酸性,催化活性差;而硅铝球载体由于氧化硅表面引入了三价铝离子,使其表面呈很强的B酸性(酸位点在金属铝氧化物的铝离子上),从而硅铝复合氧化物载体具有很强的催化活性。因此本实验催化剂载体选择硅铝球载体,其中载体尺寸为3~5mm,比表面积为273.2m/g。
1.5 臭氧催化剂制备
实验采用浸渍−焙烧法制备臭氧催化剂,具体制备步骤如下。
采用等体积浸渍的方式,将100mL 硅铝球分别浸渍至硝酸铁(浓度为1mol/L,体积100mL)、硝酸锰(浓度为1mol/L,体积100mL)、硝酸铜(浓度为1mol/L,体积100mL)、硝酸铈(浓度为1mol/L,体积100mL)的4 种溶液中,常温下置于恒温振荡器内,以150r/min 振荡24h,取出催化剂置于烘箱内,在105℃下干燥12h,最后将其置于马弗炉内焙烧。焙烧采用程序升温:升温速率以2℃/min 由常温升至150℃,恒温30min,随后以2℃/min升至300℃,恒温30min,再以5℃/min升至550℃,恒温2h,随后自然降至室温。焙烧后的催化剂即为铁基催化剂、锰基催化剂、铜基催化剂及铈基催化剂。
1.6 实验装置
反应装置如图2所示,操作流程为:氧气经流量计经反应器布气板进入水相,在催化剂作用下生成羟基自由基,从而去除废水中有机物,未反应臭氧进入尾气收集装置。实验过程中定时取样分析COD。COD去除率计算如式(1)。

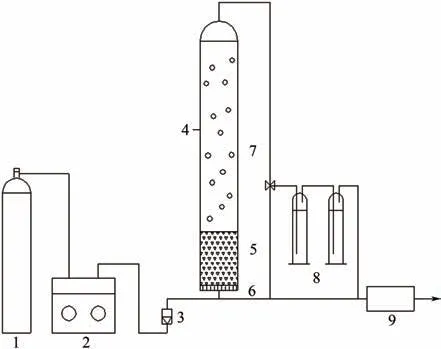
图2 臭氧催化氧化反应装置
2 结果及讨论
2.1 催化剂优选
将上述制备的4种不同活性组分的催化剂在相同条件下开展臭氧催化氧化实验。实验条件:以水样3 为实验水样,水样处理量2L/h,臭氧投加量200mg/L,催化剂投加量0.8L/L,实验60min 后取样分析COD 含量并计算COD 去除率,实验结果如图3所示。
由图3可知,不同活性组分下的催化剂对COD的去除率影响较大,负载铁、锰、铜、铈活性组分后的催化剂在反应60min 后COD 去除率分别为37.8%、36.4%、34.5%、32.3%,即活性组分硝酸铁最佳,其次是硝酸锰、硝酸铜、硝酸铈。分析原因:相比铈、铜,铁及锰的催化活性高,并且与不饱和氧原子的反应速率快,因此与臭氧反应生成羟基自由基的速率快,进而COD 去除效率高;同时铁、锰存在多种价态,金属氧化物价态变化所转移的电子有效促进臭氧分解产生·OH,有利于加快水中有机物的降解。最终选择催化剂为硅铝基−铁系催化剂。
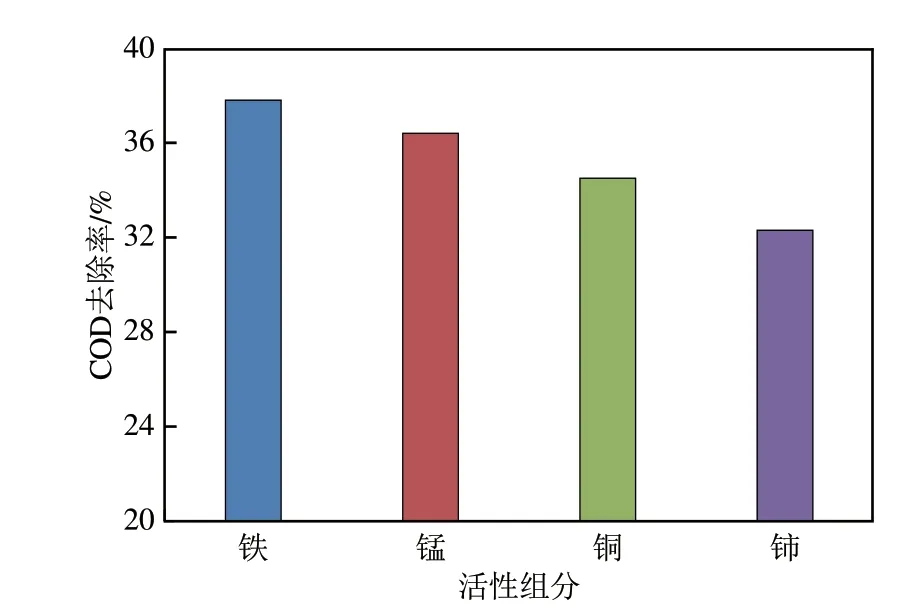
图3 活性组分种类对催化效率的影响
2.2 催化剂表征分析
采用上述方法制备硅铝基−铁系催化剂,对催化剂开展表征分析,了解催化剂的表观形貌、催化剂的元素组成、负载情况及零点电位等,为催化剂制备工艺的改进及催化剂后期的实验提供理论支撑。
2.2.1 催化剂电镜(SEM)分析
对催化剂进行SEM 分析,了解催化剂的表观形貌,如图4所示。
由图4可知,催化剂表面上分布着较多大小不等的孔。铁氧化物作为催化剂活性组分,富集在催化剂内部的孔洞内。分析原因:载体是多孔骨架结构,催化剂表面的孔和内部的孔结构相互连通,使得活性物质在浸渍过程中,由于表面张力而产生的毛细管压力作用,通过管道渗入到催化剂内部并富集在孔中。

图4 不同放大倍数下催化剂SEM图
2.2.2 催化剂X射线荧光光谱(XRF)分析
采用X射线荧光光谱对催化剂的元素组成进行分析,结果如表1所示。
如表1 所示,催化剂主要元素为Si、Al 和Fe,其次是Na、Mg和K,其中Na、Mg和K为载体的辅助成分。另外XRF点燃导致组分损失量(L.O.I)为2.87%,说明催化剂还含有少量的碳或结合水等易燃成分。根据XRF 结果分析,硅元素占到了催化剂整体质量的27.80%,同时伴有相对较高含量的铝和铁,分别占到了催化剂整体质量的11.71%和6.99%,其他金属元素(钾、钠和镁)含量较低,占催化剂整体质量的2.35%。因此通过XRF分析印证了该催化剂为多相固体催化剂,硅铝氧化物为载体,铁的氧化物作为活性组分,负载在载体表面或均匀混合于整个催化剂内部结构中。
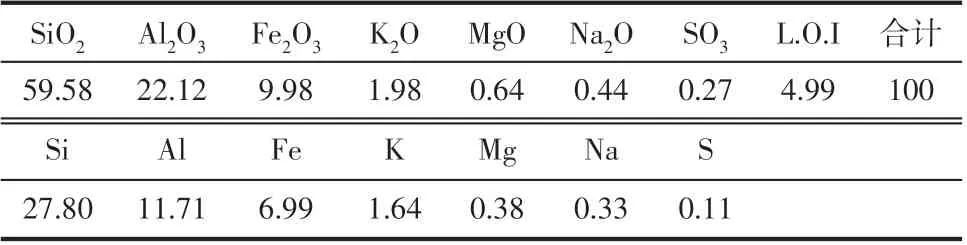
表1 催化剂元素组成分析(质量分数) 单位:%
2.2.3 催化剂截面电镜能谱分析
利用扫面电镜能谱分析仪(SEM−EDS)对催化剂截面各元素分布趋势进行分析,将催化剂机械抛光过后得到截面的切片,进行SEM−EDS 分析得到的各元素分布趋势结果如图5所示。
由图5 可知,图5(a)未发现在截面上存在任何裂缝或分界面,表明催化剂无明显“载体−活性层”结构,并且可清晰地看到多孔结构;图5(b)Fe元素集中分布于催化剂的孔洞附近,其余均匀分布于整个催化剂截面上;图5(c)~(h)分别为Si、Al、Fe、Na、Mg和K均匀分布在整个催化剂的断面上,且在孔处有缺失。其中Si 和Al 的丰度较高,是催化剂的主要元素,分布规律相近,区域基本重合。即可得出:催化剂载体为硅铝复合氧化物,铁作为活性组分均匀负载于载体上。该结果与XRF(表1)和XPS测定的结果一致。

图5 催化剂截面SEM−EDS图
2.2.4 催化剂电位表征分析
考虑在不同pH 情况下,催化剂表面电荷表现特性不同,进而影响催化剂性能(通常催化剂表面会在较高pH 条件下呈现负电,这种电性会排斥在有机介质中大部分带负电的有机物,导致催化臭氧效率降低)。实验测定了不同pH 条件下催化剂的zeta电位,结果如图6所示。
从图6 可知,催化剂零点电势在pH=3 附近,这表明大多数情况下催化剂表面带负电。分析催化剂零点电势在pH=3 附近的原因为:在SiO−AlO表面,单个铝离子会通过氧桥被三个四价硅所环绕,表面朝向缺少一个配位硅,这种不对称现象使得铝离子有强烈的亲电特性,故会吸引靠近的水分子中的负性羟基,分离出一个质子而形成较强的B酸,这种酸性中心使得硅铝复合氧化物的等电位点会出现在pH=3左右。
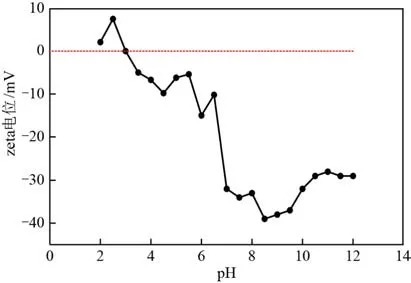
图6 催化剂zeta电位随pH变化图
2.3 臭氧催化氧化降解机理分析
为研究臭氧催化氧化降解有机物的机理,以甲酸模拟水样为实验水样,开展对臭氧催化氧化作用方式、臭氧衰减率变化、羟基自由基变化、HO变化及超氧自由基(·O)变化的影响研究。
2.3.1 臭氧催化氧化作用方式
为探讨臭氧催化氧化的作用方式。以甲酸模拟水样作为实验水样,实验过程引入叔丁醇(即TBA,其与臭氧的反应速率常数为3×10m·s,远小于与·OH 的反应速率常数6×10m·s,并且与·OH 发生反应会导致自由基链式反应终止)。实验考察叔丁醇对甲酸降解率的影响,结果如图7所示。
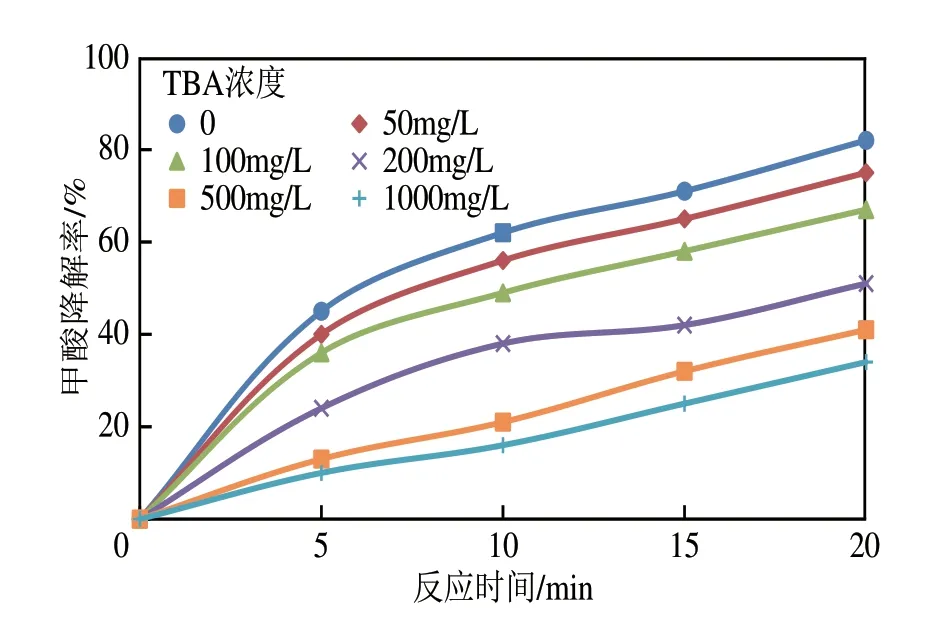
图7 不同叔丁醇浓度对甲酸降解率的影响
由图7可知,叔丁醇的加入明显抑制了对甲酸的降解效果,即在臭氧催化氧化降解甲酸的过程中,叔丁醇会与甲酸“抢占”·OH,生成惰性产物,终止自由基的链式反应,同时导致更少的·OH 与甲酸反应,进而甲酸降解效果明显降低。叔丁醇浓度为1000mg/L 时,甲酸降解效果明显受到抑制,反应时间20min 时甲酸降解率由82.3%下降至34.2%,但叔丁醇浓度大于200mg/L 时,甲酸降解率影响差别不大。原因是当叔丁醇浓度小于200mg/L 时,臭氧催化氧化过程·OH 的产生数量足以将甲酸及叔丁醇氧化,但当叔丁醇超过200mg/L时,反应产生的·OH被叔丁醇全部消耗。臭氧氧化降解甲酸过程中,除·OH 降解甲酸外,还有O直接氧化甲酸,虽然叔丁醇可抑制·OH的生成,但不能抑制O对有机物的直接氧化,因此当叔丁醇浓度为1000mg/L 时,甲酸仍有一部分被降解。
综上所述,叔丁醇具有抑制·OH 的作用,同时叔丁醇的加入明显降低了催化氧化过程中甲酸的降解效率,因此可证明臭氧催化氧化遵从羟基自由基的作用机理,即臭氧在催化剂表面活性位点化学吸附生成羟基自由基,羟基自由基进而降解水中有机物。
2.3.2 反应体系羟基自由基变化规律影响
臭氧降解甲酸的过程包括两个方面,即直接反应和间接反应。其中间接反应为O分解产生·OH,与甲酸发生反应。为考察实验过程中羟基自由基的变化规律。以5,5−二甲基−1−吡咯啉−−氧化物(DMPO)为羟基自由基捕获剂,检测反应10min时臭氧氧化、臭氧/载体(O/AT)、臭氧/催化剂(O/MAT)体系下·OH信号强度,如图8、图9所示。
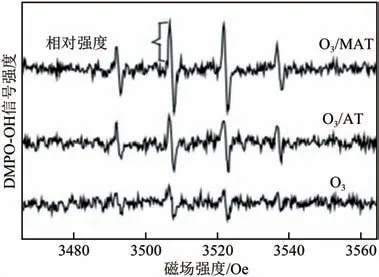
图8 不同体系下·OH信号强度对比
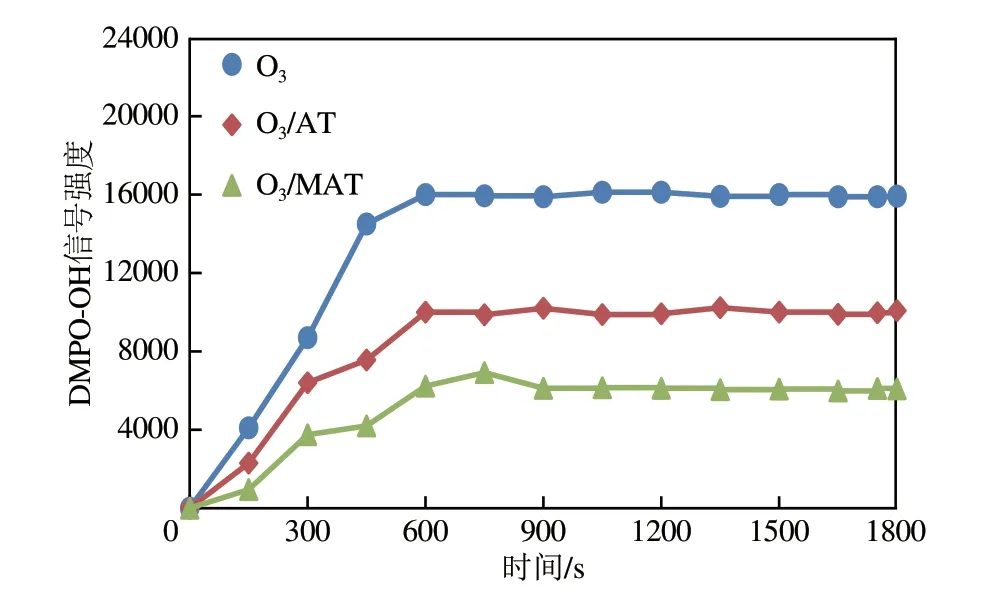
图9 不同体系下·OH信号强度随时间变化
由图8 可知,臭氧、臭氧/载体、臭氧/催化剂体系均能生成·OH,同时臭氧/载体及臭氧/催化剂的·OH 信号强度大于臭氧体系,此结果进一步表明臭氧氧化及臭氧催化氧化过程均遵循·OH 作用机理理论。
由图9 可知,反应前10min,臭氧、臭氧/载体、臭氧/催化剂体系的·OH信号强度随时间延长而增加,反应速率分别为11.23A.U./s、18.94A.U./s和30.46A.U./s。即O不断溶解水中进行传质的过程中,·OH生成数量逐渐增大,反应10min后生成数量趋于稳定状态。臭氧/催化剂·OH的生成数量远大于臭氧及臭氧/载体,这表明臭氧催化剂的加入促进了·OH 的生成速率及数量,进而更加快速地降解水中有机物。
2.3.3 反应体系HO变化规律影响
HO是臭氧催化氧化过程中可能产生的氧化剂,可与羟基自由基发生反应从而相互转化及淬灭,有必要探讨臭氧催化氧化过程中HO的生成及变化。实验对比了臭氧、臭氧/载体、臭氧/催化剂在pH为3、7.3、8.5下HO的产生情况,结果如图10所示。
由图10可知,臭氧、臭氧/载体、臭氧/催化剂体系在不同pH 下HO的产生量均会在短时间内达到稳定值,即HO作为臭氧催化氧化降解体系中的中间产物,将在短时间内达到生成及分解平衡。同时臭氧/催化剂下HO生成量虽高于臭氧及臭氧/载体的HO生成量,但并未因催化剂的加入导致HO生成量大幅增加。在pH=3 时,HO生成量随时间延长而逐渐增加;而在pH=7.3及8.5时,HO生成量在10min 内达到最大值。不同pH 下,臭氧及臭氧/催化剂在10min 时HO生成情况与图11 中·OH类似,初步猜测HO的生成与·OH有关。
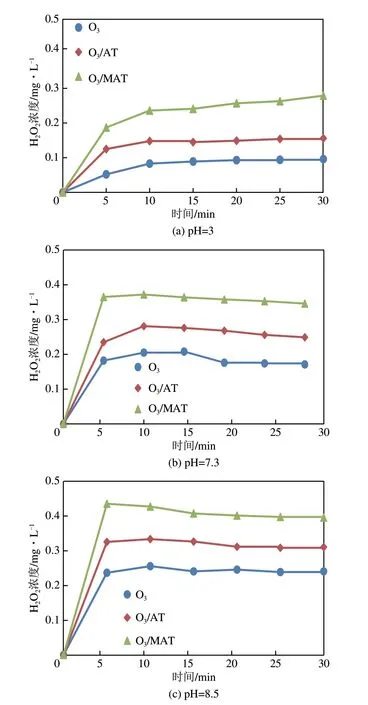
图10 不同pH下H2O2浓度变化
为进一步推测HO与·OH 的关系,在实验过程中引入叔丁醇,结果如图11所示。
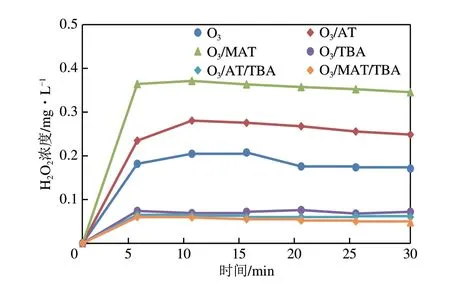
图11 不同体系下叔丁醇对H2O2生成量的影响
由图11 可知,叔丁醇抑制了反应过程中HO的生成,由此表明HO的平衡浓度和·OH 生成有关。在引入叔丁醇30min后,臭氧体系下HO的生成量由0.164mg/L 降低至0.085mg/L,臭氧/载体体系下HO的生成量由0.228mg/L 降低至0.064mg/L,臭氧/催化剂体系下HO的生成量由0.342mg/L降低至0.054mg/L,即在叔丁醇存在下,臭氧/催化剂体系的HO生成量最小,而减少幅度最大。这表明·OH 对HO的生成具有重要作用,即臭氧/催化剂体系下·OH 的生成量远大于其他两个体系,而·OH 被叔丁醇抑制后生成大幅下降,进而由·OH生成HO的数量便会降低。
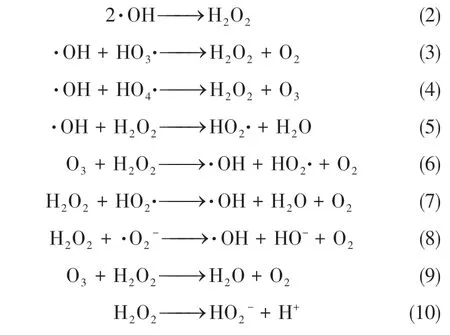
由于HO和·OH 可通过发生反应相互转化及淬灭,因此反应过程中HO和·OH 存在平衡状态,推测反应历程如式(2)~式(10)。
2.3.4 反应体系超氧自由基变化规律影响
超氧自由基(·O)是臭氧催化氧化过程中可能产生的氧化剂,有必要探讨臭氧催化氧化过程中(·O)的生成及变化。实验对比了臭氧、臭氧/载体、臭氧/催化剂在pH 为3、7.3、8.5 下HO的产生情况,结果如图12所示。
由图12可知,臭氧、臭氧/载体、臭氧/催化剂在不同pH下·O的产生量均会在短时间内达到稳定值。在pH=8.5 时,·O产生量明显高于pH=3 和pH=7.3 时的·O产生量,原因是在碱性条件下,溶液中含有大量的OH,从而可促进O快速分解生成大量·O。
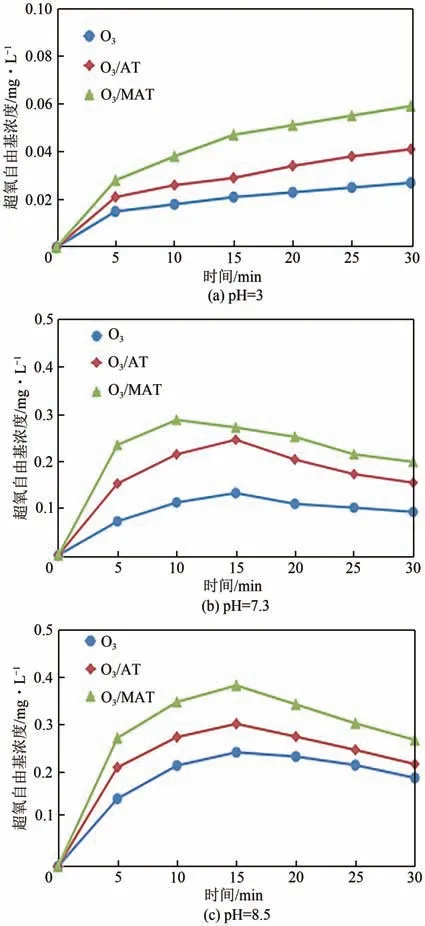
图12 不同pH下·O2−浓度变化
为进一步推测·O与·OH的关系,在实验过程中引入叔丁醇,结果如图13所示。
由图13 可知,TBA 的引入对·O生成量影响不明显,即叔丁醇虽抑制了·OH的产生,但不会抑制·O的产生,因此在臭氧催化氧化过程中·O的产生与·OH没有关系。
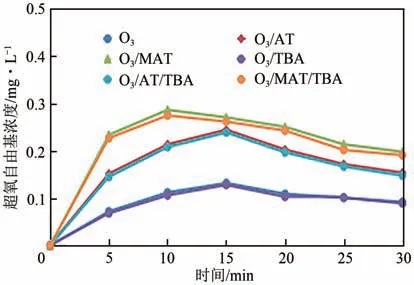
图13 不同体系下TBA对·O2−浓度变化
2.3.5 催化剂对臭氧衰减的影响
以甲酸模拟水样开展臭氧氧化及催化臭氧氧化实验,对反应前后水样中臭氧识别表征,结果如图14所示。
由图14可知,在pH=3下,催化剂对臭氧衰减的促进作用更明显,这与催化剂较低的pH(=3)有关。而在pH=7.3 和pH=8.5 的高pH 条件下,绝大部分臭氧通过自衰减实现衰减。其中主体溶液中臭氧浓度降低可主要归因于以下两点。
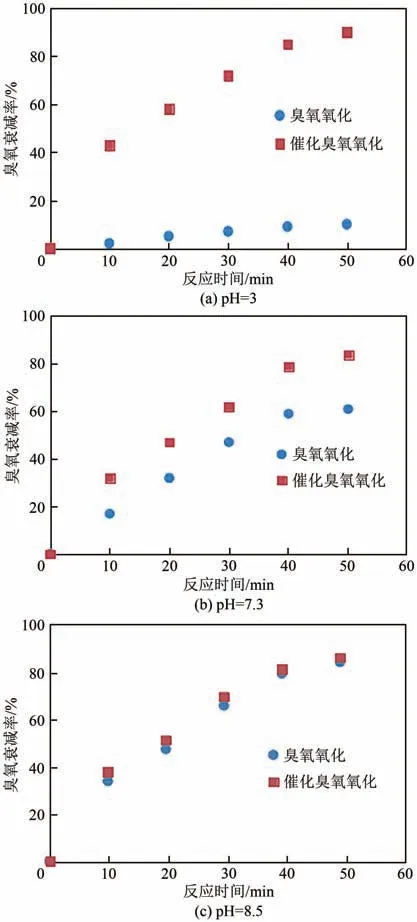
图14 不同pH下催化剂对臭氧衰减率的影响
(1)臭氧被吸附到催化剂表面活性位点,促进臭氧催化分解生成羟基自由基。羟基自由基可被催化剂表面物理吸附或扩散回主体溶液。
(2)臭氧被催化剂无用消耗,不产生额外的氧化剂。
考虑臭氧催化氧化过程为羟基自由基作用机理,结合臭氧在水相中分解产生HO、HO·、HO·、HO·、·O、·O及·O等,可推测反应过程中·OH的产生历程如式(11)~式(17)。
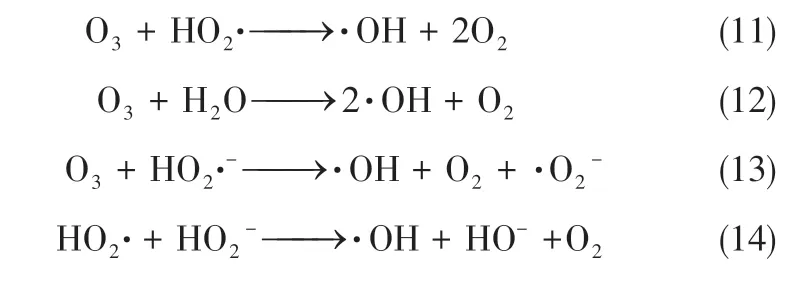
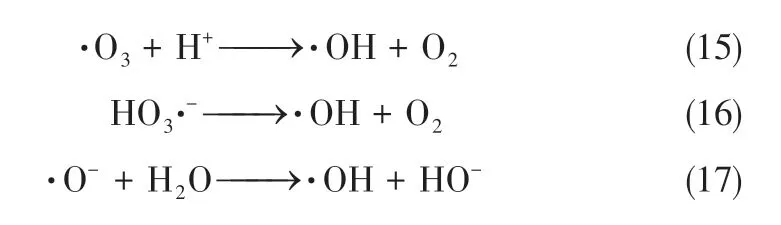
3 结论
通过对煤化工高盐废水臭氧催化剂表征分析与降解机理研究,得到以下结论。
(1)对国内三种典型煤化工高盐废水进行水质分析,煤化工高盐废水中阴阳离子种类及含量随工艺不同而各不相同,但三种煤化工高盐废水阳离子主要为钠离子,还含有少量的钾离子、钙离子及镁离子;水中阴离子主要为氯离子、硫酸根,其次为硝酸根离子。
(2)通过对催化剂活性组分开展臭氧催化氧化试验研究发现,活性组分对臭氧催化氧化效率影响较大,考虑氧化效率最终选择催化剂为硅铝基−铁系催化剂。
(3)通过对硅铝基−铁系催化剂进行内部及表面表征分析可知:①通过对催化剂表面电镜分析,可知催化剂表面活性组分负载均匀;②通过对催化剂整体XRF 分析,印证了该催化剂为多相固体催化剂,硅铝氧化物为载体,铁的氧化物作为活性组分,负载在载体表面或均匀混合于整个催化剂内部结构中;③通过对催化剂截面SEM−EDS 分析,可知催化剂载体为硅铝复合氧化物,铁为活性组分均匀地负载于载体上,同时该结果与XRF结果一致;④通过对催化剂电位表征可知,催化剂的零点电势在pH=3附近。
(4)通过对臭氧催化氧化机理进行研究可知:①臭氧催化氧化过程遵从羟基自由基作用机理,即臭氧在催化剂表面活性位点化学吸附生成羟基自由基,羟基自由基进而降解水中有机物,同时臭氧催化剂的加入促进了·OH 的生成速率及数量;②臭氧催化氧化过程中HO生成量与·OH 有关,·OH越多,HO生成量越多,但·O−的产生与·OH没有关系;③臭氧催化氧化过程中臭氧通过衰减产生羟基自由基,其中臭氧衰减的原因:一是臭氧被吸附到催化剂表面,导致臭氧表面官能团反应生成羟基自由基,二是臭氧被催化剂无用消耗,不产生额外的氧化剂。
