IQSEC2基因变异相关的癫痫性脑病1例报告
2022-02-10郎长会田茂强雷文婷李娟束晓梅余小华
郎长会,田茂强,雷文婷,李娟,束晓梅,余小华
癫痫性脑病(epileptic encephalopathy,EE)是一种严重的顽固性癫痫发作和神经功能障碍性疾病,其中癫痫活动本身可能导致严重的神经和认知功能损害[1]。随着二代测序的广泛开展,EE的致病基因越来越多。其中,IQSEC2(IQ motif and SEC7 domain containing protein 2)基因变异最早于2008年在1例婴儿痉挛中被发现[2]。IQSEC2基因位于Xp11.22染色体上,在大脑和脊髓中表达,并在神经元、细胞骨架、树突及兴奋性突触中发挥关键作用。IQSEC2基因具有多个重要功能结构域,其基因变异与X连锁EE和X连锁智力低下、难治性癫痫、孤独症(autism spectrum disorder,ASD)及Lennox-Gastaut综合征(Lennox-Gastaut syndrome,LGS)等有关,目前全球约有140余例IQSEC2基因变异相关患者[3-5]。国内仅有个案报道IQSEC2基因变异与X连锁精神发育迟滞有关,但未显示有癫痫表型[6]。本文回顾性分析1例以发育落后为表现的难治性癫痫患儿的临床资料,以提高临床医师对该基因变异特点的认识。
1 病例报告
患儿,男,3岁零6个月,因自幼发育落后,间断抽搐1年,加重1个月入院。患儿8个月时能抬头数秒,1+岁不能独坐,2+岁不能独站,3+岁不能独走,病后有发育倒退现象,现不能抬头、独坐,不会说话,少有目光交流,对叫名无反应,喜搓手,认知、理解差,伴喂养困难。1年前出现反复抽搐,有5种发作形式:(1)表现为突发双目凝视,呼之不应,动作减少,持续数秒后缓解,缓解后玩耍自如,家属未予特殊重视,现已无此种发作;(2)1个月后出现点头、呈单下发作,平均每天1~3次,多时每天7~20次不等;(3)表现为偶尔头下垂,伴或不伴身体前倾;(4)表现为双眼凝视(偶尔为突发尖叫),呼之不应,伴唇周发绀、四肢强直、抖动,时有口吐白沫或流涎,持续数十秒至3~4分钟后予止惊或自行缓解,现无此种发作;(5)表现为双目凝视,嘴角抽动、眼角眨动,持续数秒可缓解,现无此种发作。家族史:母亲有智力障碍,能独立行走,能简单交流,生活勉强能自理,幼时抽搐发作1次,未予特殊处理。父亲有轻度智力障碍,生活能自理。个人史:患儿系第1胎第1产,出生时否认缺氧及窒息史。查体:无特殊外貌及色素胶失斑,心肺腹查体无特殊,四肢肌张力低下,病理征阴性。实验室检查:血常规、生化无特殊,血氨、乳酸及同型半胱氨酸正常。中国儿童发育量表[DST测试(3岁时)]:发育商(DQ)<50分,智力指数(MI)<48。Gesell发育量表(4岁时)评估:智力发育相当于3个月左右。视频脑电图为异常儿童脑电图,表现为背景枕区节律少,节律慢化;清醒期双侧额、中央、顶、枕、颞区及中线区δ慢波发放;以双侧后头部或双侧后头部为主的广泛性(左侧著)棘波、尖慢及多棘慢波发放。2岁零6个月时头颅MRI未见明显异常。复查视频脑电图示:(1)监测到清醒期1次痉挛发作(图1A),大量双侧后头部为主的多灶性棘波、棘慢波、尖慢波、多棘慢波及慢波发放;(2)睡眠期大量广泛性1.5 Hz~3 Hz棘慢波发放(图1B)。患儿3岁零6个月时头颅MRI示轻度脑萎缩。孤独症评定量表评估:患儿存在明显的孤独症症状,表现为与人交流非常困难,几乎无法进行语言或非语言的沟通,运用语言能力极差,不能提出话题或维持话题,非语言交流缺乏,对特殊的物品或活动不感兴趣。患儿的视频脑电图见图1。
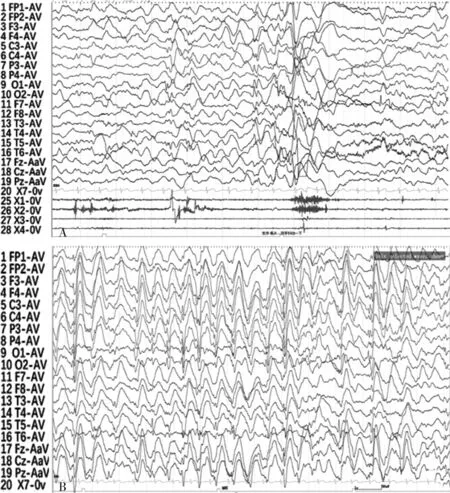
注:A为监测到一次痉挛发作,B为睡眠期大量广泛性1.5 Hz~3 Hz棘慢波发放。图1 患儿的视频脑电图
经过家属知情同意及遵义医科大学附属医院伦理委员会批准(伦理号:KLL-2022-044),外送家系全外显子组测序检测发现患儿IQSEC2c.424C>T (p.Gln 142*) (NM_001111125.3)半合子变异(图2),该变异来自智力障碍的母亲,经ACMG评级为可疑致病性变异。PVS1:该变异为无义突变;PM2:该变异在ExAC、gnomAD及千人数据库中没有收录;PP4:该基因变异症状与患儿临床表现相符。该变异位点在国内外目前鲜有报道。该基因关联疾病为X连锁智力障碍、癫痫及ASD等。患儿为半合子,母亲为杂合子(母亲有智力障碍),遗传模式可解释患儿临床表现。
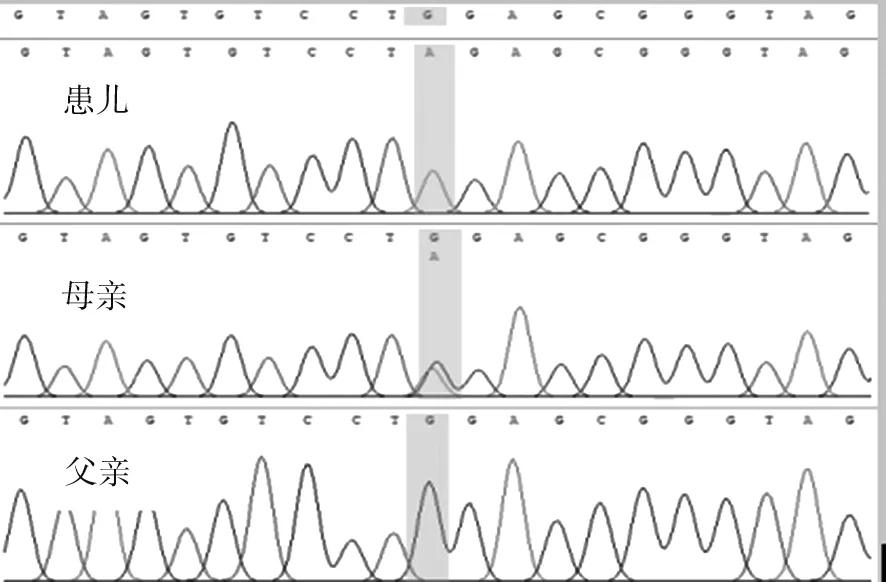
注:患儿在外显子发生碱基无义变异[(c.424C>T (p.Gln 142*)],该变异来源于智力障碍的母亲,父亲未发生变异。图2 患儿及其父母的IQSEC2基因测序图
根据患儿临床表现及实验室检查诊断为难治性癫痫,其发作形式主要为全面强直阵挛发作、失神发作、痉挛发作、失张力发作及局灶性发作(IQSEC2基因变异相关),先后小剂量口服左乙拉西坦、丙戊酸钠及托吡酯片,曾因痰多而停用氯硝西泮。8个月前就诊于外院,停用托吡酯片,改为唑呢沙胺片[120 mg/d,约10 mg/(kg·d)];左乙拉西坦[500 mg/d,约41.6 mg/(kg·d)],丙戊酸钠[480 mg/d,约40 mg/(kg·d)],入院后予甲泼尼龙冲击治疗后患儿痉挛发作减少,现激素减量治疗过程中。末次随访为4岁零2个月,发育无明显进步,目前每天仍有4~5次点头痉挛发作,少则2~3次,多则6~8次;失张力发作为每天3~5次不等(发作形式变化见图3)。

图3 患儿的临床发作变化图
2 讨论
IQSEC2基因也称为BRAG1基因[7,10-11],长度约93.7 kb,主要有2种mRNA异构体:第1种异构体包含15个外显子(GenBank:NM_001111125.3)及1 488个氨基酸(NP_001104595);第2种异构体包含14个外显子(GenBank:NM_015075.1)及949个氨基酸(NP_055890)。IQSEC2蛋白又称为鸟嘌呤核苷酸交换因子(guanine-nucleotide exchange factor,GEF),该蛋白是一种多结构域蛋白,包括N端螺旋-螺旋域、Sec7结构域、IQ 钙调蛋白结合模块、Pleckstrin结构域及C端PDZ结合功能域[8]。IQSEC2蛋白可以激活ADP核糖基化因子6 (ADP ribosylation factor,ARF6),其对于维持正常学习所必需的兴奋性和抑制性受体水平是极其重要的[9]。IQSEC2 mRNA在嗅球、大脑皮层、海马及小脑中高表达,其蛋白产物位于兴奋性突触致密物质上。IQSEC2和突触后致密物蛋白通过与C端PDZ结合相互作用,与N-甲基-D-天冬氨酸(N-methyl-D-aspartate,NMDA)受体形成复合物,在α-氨基-3-羟基-5-甲基-4-异恶唑烯丙酸(α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid,AMPA)受体的运输中发挥作用,从而在神经元发育和突触可塑性中发挥重要作用[10--11]。IQSEC2基因通过突触后致密物-95与NMDA型谷氨酸受体结合,NMDA受体的激活导致Ca2+内流并与IQSEC2 IQ结合模块上的钙调蛋白结合,Ca2+/钙调素诱导IQSEC2的构象变化,导致Sec7催化域的激活,促进表面AMPA型谷氨酸受体下调[9-10]。突触传递中的兴奋和抑制之间的不平衡被认为是IQSEC2基因变异引起ASD的病理生理学基础。
目前文献报道的140余例IQSEC2基因变异患者中至少有70种不同的变异,包括无义变异、移码变异、剪接变异、缺失及错义变异等。这些变异都与中度至重度智力障碍、各种癫痫发作及ASD特征有关。IQSEC2基因变异相关的临床表型在性别方面差异性较大,男性临床特征包括肌张力减退、中至重度精神运动发育迟缓、智力低下、难治性癫痫、言语障碍、ASD样特征及刻板动作等[10-17]。本例患儿发育落后严重,有多种癫痫发作形式且对多种抗癫痫药疗效欠佳,伴有ASD,结合脑电图有1.5 Hz~3 Hz棘慢波发放,需警惕向LGS转型,且既往文献报道突触传递相关IQSEC2基因变异可导致LGS或LGS样综合征,此与文献相符。在杂合子女性患者中多数表现为轻度智力低下,少数有严重智力低下,多伴有癫痫发作(10/19)[10],但男性发育迟缓、智力低下及癫痫发作均比女性严重[9]。Migot等[11]比较18例男患和19例女患IQSEC2基因变异临床表现的差异,发现女性致病性变异全是新发变异,而男性半合子变异可为新发,也可来自母亲。由于IQSEC2基因定位于X染色体上,女性携带者的临床表现可从无症状携带者到轻度智力低下,且女性症状通常比男性轻。因为女性IQSEC2基因可发生X染色体非随机失活[10],但该基因也可逃避X染色体非随机失活,这可解释在杂合子女性中智力低下及ASD患病率较高的原因[9]。本例患儿母亲为杂合子,其症状相对于患儿来说相对较轻,有智力障碍,在幼时曾有惊厥发作,此与文献报道相符。Ewans等[15]报道一家系中4例女孩有发育迟缓(中度到重度智力障碍),在2~5岁时出现癫痫发作,以局灶性发作为主,发病诱因包括发热及免疫力低下等,幼时有ASD及攻击性行为,其中两例女孩在青春期出现发育倒退,分别在16岁和22岁时死于癫痫猝死。因此需嘱咐其监护人在看护过程中预防突发猝死。
IQSEC2基因变异相关患者的头颅MRI多数正常(21/36),少数可出现非特异性异常,包括轻度脑萎缩、胼胝体薄及髓鞘形成延迟等[9]。本例患儿早期头颅MRI正常,后期复查出现轻度脑萎缩,此与文献相符。
80%以上IQSEC2基因变异相关的癫痫是难治性的。Izumi 等[18]报道IQSEC2基因变异相关的癫痫以顽固性、早发癫痫性痉挛和强直/强直阵挛性发作为特征,经多种抗癫痫药物包括糖皮质激素及VPA等治疗均欠佳。Choi等[5]用左乙拉西坦、丙戊酸、唑尼沙胺、鲁非胺及氯巴占等抗癫痫药物治疗IQSEC2基因变异引起的癫痫,均无效,患者每月仍有癫痫发作。本例患儿经多种抗癫痫药物治疗后尽管痉挛发作频率减少,但每天仍有癫痫发作,此与文献报道相符。
综上所述,IQSEC2基因变异引起的癫痫在男性中多为难治性,智力低下多为中至重度,且多伴ASD等,在杂合子女性中智力低下多为轻度至中度,癫痫发作频率较少。因此需要进一步在动物模型中研究IQSEC2基因变异在男性患儿中的发病机制,以提高此类患儿癫痫发作的缓解率。
